

You are here: Home > Section on Macromolecular Recognition and Assembly
Intermolecular Forces, Recognition, and Dynamics

- Donald C. Rau, PhD, Head, Section on Macromolecular Recognition and Assembly
- James M. Hutchison, BA, Postbaccalaureate Intramural Research Training Awardee
- James Ritchie, BA, Postbaccalaureate Intramural Research Training Awardee
Our goal is to elucidate the coupling of the thermodynamics with structure of biologically important complexes and assemblies. The next challenge in structural biology is to understand the physics of interactions between molecules in aqueous solution. The ability to take advantage of an increasing number of available protein and nucleic acid structures will depend critically on establishing the link between structures and interaction energies. A fundamental and quantitative knowledge of intermolecular forces is necessary to understand the strength and specificity of interactions among biologically important macromolecules that control cellular function and to rationally design agents that can effectively compete with such interactions when associated with disease. Our results have shown that, when two molecules are very close, experimentally measured forces are different from those predicted by current, conventionally accepted theories. We have interpreted the observed forces as indicating the dominating contribution from water-structuring energetics.
Our practical goal is to apply the lessons from direct force measurements to the recognition and assembly reactions that control cellular processes. DNA assembly and compaction is a universal feature of cell biology. We are investigating two systems in which DNA is packed to liquid crystalline densities. DNA in vertebrate sperm nuclei is organized differently from any other eukaryotic cell. DNA is spontaneously compacted into closely packed hexagonal arrays by arginine-rich peptides called protamines. Given that DNA repair is absent from sperm, it is thought that dense packaging of DNA in sperm nuclei is necessary to protect the genome against damage by mutagens and reactive oxidizing species (ROS). DNA damage has been linked to infertility and likely contributes to miscarriages and birth defects. Our knowledge of intermolecular forces, in general, and DNA packing, in particular, helps elucidate the protamine dysfunctions that cause DNA packaging defects in sperm. The DNA in several viruses is also close packed, but such helices are strongly repulsive. We have been investigating the impact of closer packing on DNA injection and consequent infection.
DNA packaging in sperm nuclei
Protamines are small (30–50 amino acids), arginine-rich peptides that are used to package DNA densely in vertebrate sperm nuclei. Protamines spontaneously assemble DNA from dilute solution into extended, close-packed (less than 10 Å surface-to-surface separation), liquid crystalline arrays that are suitable for characterization by X-ray scattering. Reconstituted protamine-DNA assemblies and sperm nuclei give very similar X-ray scattering profiles. Using synthetic peptides, we previously determined that (i) protamines use arginine almost exclusively over lysine to condense DNA because lysine-rich peptides package DNA very poorly by comparison and (ii) the DNA packing density in salmon sperm nuclei is entirely determined by the amino acid content of salmon protamine, i.e., 21 arginines and 10 neutral amino acids.
Salmon sperm nuclei can be fractionated on a sucrose density gradient. The denser fraction has a narrow distribution of DNA interhelical distances with a peak at around 9 Å surface-to-surface separation. The less dense fraction has the same peak maximum, but the scattering profile is skewed toward larger interhelical separations, indicating a DNA subpopulation that is more loosely packed. Adding excess protamine to the light fraction recovers the scattering profile of the denser fraction. The skewed scattering profile from less dense sperm nuclei is a consequence of insufficient protamine.
Insufficient protamine affects DNA packing in reconstituted protamine-DNA assemblies differently from that in sperm nuclei. Reconstituted assemblies with only 90–95% of the protamine necessary to neutralize DNA show a scattering profile that simply shifts the scattering peak to a larger interhelical distance rather than skewing the scattering profile. We hypothesize that protamines can equilibrate throughout a reconstituted condensate, but not in sperm nuclei, which would suggest that the DNA in sperm nuclei is organized into discrete domains that hinder protamine equilibration.
The (101) X-ray reflection is a combination of interaxial and helical repeat scattering. Its amplitude strongly depends on the correlation of backbone phosphates on adjacent helices. There is a distinct difference in the intensities of the (101) reflections relative to the main interaxial reflection between reconstituted assemblies and sperm nuclei. The relative loss of intensity of the (101) reflection with nuclei is consistent with the suspected toroidal packing of DNA in sperm nuclei. Toroidally constrained DNA is strongly curved, disrupting interhelical phosphate-phosphate correlations. The suspected toroids may also be the domains that hinder the equilibration of protamine throughout the nucleus. We are probing a possible domain structure using nuclease digestion.
Protamines are initially serine-phosphorylated when replacing histones on DNA. Dephosphorylation during spermiogenesis is coupled to compaction of the nucleus. Incomplete dephosphorylation has been linked to DNA damage. About 1 % of salmon protamines isolated from sperm nuclei are still phosphorylated. We previously found that serine phosphorylation of synthetic arginine peptides greatly reduces the attraction between helices, far more than would be expected based on a simple reduction in net charge. Herring protamines can be partially phosphorylated with protein kinase A. We found that protamine phosphorylation does dramatically reduce the attraction between helices (Figure 1) in reconstituted DNA assemblies. Even modest phosphorylation that reduces the protamine charge by only 5% increases the aqueous volume accessible to oxidizing species by 50% in reconstituted assemblies. We can extrapolate that fully phosphorylated protamines would still bind to DNA quite strongly, but would be unable to condense DNA into tightly packed assemblies. We hypothesize that a systematic and sequential dephosphorylation of protamine during spermiogenesis allows for an orderly compaction of DNA.
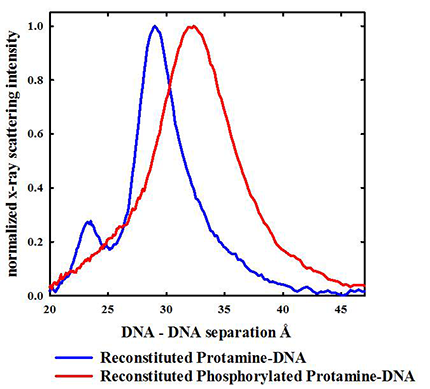
Click image to enlarge.
Figure 1. Even partial phosphorylation of herring protamine has a dramatic effect on DNA packing.
X-ray scattering profiles are shown for spontaneously condensed DNA assemblies reconstituted with either normal herring protamine or protein kinase A (PKA) serine-phosphorylated herring protamine. Only about half the PKA–reacted protamines are phosphorylated on a single serine compared with the 21 arginine charges per peptide. Even though the protamine is only very modestly phosphorylated with a 5% decrease the average charge, the average interhelical distance increases from 29.5 Å to 32.5 Å, and the scattering peak significantly broadens. Given the DNA helix diameter of about 20 Å, the increase peak in spacing accounts for a 50% increase in the aqueous volume accessible to oxidative species that can damage DNA. The peak at 23 Å seen for the reconstituted protamine-DNA sample is the (101) reflection that is indicative of ordered interhelical DNA phosphate-phosphate correlations. Such ordering is lost when DNA is condensed by phosphorylated protamine, again indicative of a much weaker attraction.
Many mammals have two protamines, P1 and P2, which are both longer than piscine protamines, about 50 vs. 30 amino acids. We are now investigating DNA packaging in bull sperm. Bull sperm only have a P1 protamine and represent an intermediate in complexity between fish and human sperm. A major difference between piscine and mammalian protamines is that extensive intra- and inter-protamine disulfide bridges are present in mammalian sperm. Bull protamine P1 contains seven cysteines, whereas salmon protamine has none. Four of the cysteines in bull protamine are involved in intra-peptide disulfide bonds and the other three in inter-protamine disulfide bonds. The intra-protamine disulfide bonds form hairpins at both ends and result in an effective linear arginine charge density that is about the same as for salmon. X-ray scattering profiles from bull sperm nuclei establish that the packing density of DNA is about the same as for salmon sperm nuclei but that the packing is less ordered. The interaxial reflections are broader, and no (101) peak is observed. Reduction of disulfide bonds with DTT has interesting consequences for order. Reduction of the more accessible inter-protamine disulfide bonds results in a much narrower interaxial X-ray scattering peak with an emergent (101) peak. The DNA relaxes to a more ordered state. The inter-protamine disulfide bonds appear to hold helices in a constrained, non-optimal conformation. Further reduction of the intra-protamine disulfide bonds leads to significant swelling of nuclei, with the release of protamine tails and consequent decrease in the effective linear density of arginines below the level needed for strong spontaneous assembly. Nuclei can still be held together and interhelical spacings measured, using an osmotic stressing agent such as polyethylene glycol to constrain swelling.
Temperature-mediated DNA packing transitions in viruses facilitate infection.
DNA is tightly packaged in a spherical spool within the protein capsids for both the bacteriophage lambda and human Herpes simplex virus type 1 (HSV-1). The DNA in the viruses is packed to liquid crystalline densities. Unlike sperm nuclei, DNA is forced into the viral capsid by strong molecular motors. Helices are strongly repulsive, and the sealed capsid is under enormous pressures. Such pressures are considered necessary to begin the process of injecting DNA into a cell. Given that DNA is a stiff polymer, the DNA in the center of the capsid is also under considerable bending stress. In collaboration with Alex Evilevitch, we uncovered a novel, temperature-mediated transition in DNA packing for both viruses. Both X-ray scattering and cryoEM reconstruction indicate a transition in DNA packing that occurs at about 30o C and is characterized by a substantial increase in the amount of disordered DNA in the center of the capsid (Figure 2). Concomitant measurement of whole virus compressibility, using atomic force microscopy (AFM), indicates that the virus is much softer or more fluid-like at the higher temperatures. The fact that the transition occurs close to the natural infection temperature for both viruses suggests a biological significance. Although the ejection of DNA from capsids is driven by a sizable repulsive hydration force between helices, the frictional hydration force from dragging ordered helices past one another would greatly slow the ejection rate. We hypothesize that the more disordered packing of the central helices leads to a more fluid-like state of the DNA, facilitating ejection of DNA into the target cell. A similar transition is not seen for DNA condensed from bulk solution. We thus postulate that the DNA bending energy from being condensed within a spherical protein capsid drives the transition. The softening transition that occurs at higher temperatures leads to the assumption that local DNA melting or kinking underlies the increase in disorder.
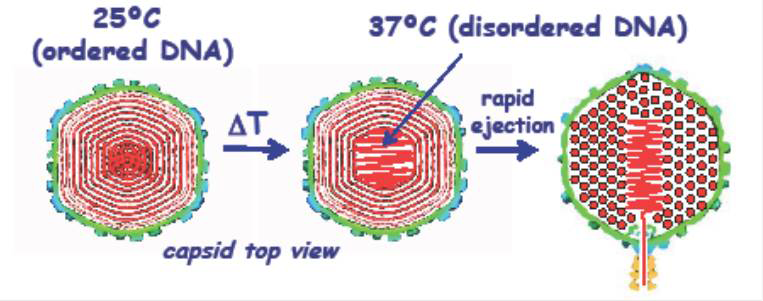
Click image to enlarge.
Figure 2. A schematic representation of the temperature-mediated transition in DNA packing observed for both bacteriophage lambda and human Herpes simplex virus-1
The DNA in the viral capsid heads of bacteriophage lambda and human Herpes simplex virus 1 is densely packed in a spool with local hexagonal symmetry. The DNA is sufficiently ordered that X-ray scattering can be used to characterize the packing. At room temperature, both X-ray scattering and cryoEM reconstruction show that only a small fraction of the DNA is disordered in the capsid center. A transition to a state with much more centrally located, disordered DNA, however, is observed at around 30o C, about at the temperature of infection. Atomic force microscopy establishes that the transition is accompanied by a large reduction in the compressibility of the viral capsid. We postulate that the central disordered DNA is fluid-like, enabling faster ejection.
Relating hydration force amplitudes and surface chemistry
Our direct measurement of forces between several markedly different macromolecules—charged, zwitterionic, and wholly uncharged—showed that water structuring dominates interactions at close distances, the last 10–20 Å separation. The hydration forces show two components: a longer-range exponential interaction with a decay length of about 4–5 Å that is attributable to the direct interaction of the hydration structure on one surface with that on an adjacent surface. The force can be either repulsive or attractive, depending on the extent of complementarity of water structuring on the two interacting surfaces. A second exponential force is always repulsive and has half the decay length, about 2–2.5 Å. This force is the hydration equivalent of an electrostatic image charge repulsion. Applied to DNA, force amplitudes necessarily reflect not only the hydration of the DNA itself but also the water structuring around DNA–condensed counterions. We previously parsed the amino acid contributions to the packing density of protamine-DNA assemblies. Using synthetic peptides, we uncovered relatively simple rules that govern the force amplitudes. The long-range attraction increases with the number of arginines in the peptide, the force amplitude varying linearly with the inverse of the number of arginines. Such dependence is consistent with the expected interplay between correlations of charges on adjacent helices and the associated peptide entropy loss. The short-range repulsion, however, is only slightly dependent on the number of arginines. By contrast, the presence of neutral amino acids increases the short-range repulsion while minimally affecting the attraction. We postulated that neutral amino acids displace water from the DNA without significantly affecting the arginine-DNA phosphate attraction. The displaced water would increase the hydration image charge-like repulsion.
To further connect structure, hydration, and force amplitudes, we investigated a set of homologous mono- and divalent alkylamine cations. Using the force curve of NH4+–DNA as the base, the addition of a single methyl group (H3N+-CH3) increases the long-range repulsion without much effect on the short-range, hydration image–like repulsion. The substitution reduces the number of hydrogen bonds the counterion can make with water and therefore reduces hydration of the ion. Adding a second alkyl carbon to the first (H3N+-CH2-CH3) has almost no effect on the long-range repulsion, but significantly increases the short-range, 2.5 Å decay-length repulsion. Addition of another carbon group [H3N+-(CH2)2-CH3] follows the same trend. The number of hydrogen bonds that can be made with water remains constant, but waters are displaced from DNA by the alkyl groups, increasing the image-like repulsion. Adding additional methyl groups directly to the nitrogen [H2N+(CH3)2 and N+(CH3)4] both reduces the number of hydrogen bonds the cation can make with water, and therefore the water structuring around these ions, and displaces water from DNA. Both force amplitudes increase significantly. With the monovalent alkylamines, the longer-ranged force simply depends on the number of hydrogen bonds the amine can make with water, whereas the 2.5 Å decay-length force is sensitive primarily to the number of alkyl groups.
While the monovalent alkylamines approximately follow the trend expected from the arginine-neutral amino acid series, the divalent alkylamines show a different behavior. We investigated the series NH2+-(CH2)M-NH2+ for M ranging from 2 to 5. The M = 4 and 5 diamines are the biogenic amines, putrescine and cadaverine, respectively. The force curves are shown in Figure 3. In this case, the alkyl linker length has almost no effect on the short-range 2.5 Å decay-length force, but the repulsive amplitude of the longer-ranged force increases linearly with linker length. The difference in the effect of alkyl groups between mono- and divalent alkylamines illustrates that force amplitudes depend sensitively not only on the chemical nature of the counterion but also on its binding mode, i.e., territorial, phosphate, or groove.
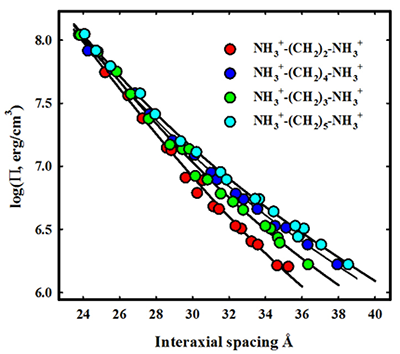
Click image to enlarge.
Figure 3. Linking structure and forces: dependence of alkyl diamine DNA forces on linker chain length
The osmotic pressure of an excluded polymer such as polyethylene glycol applies a force on a macroscopic ordered array of DNA helices neutralized by various divalent alkyl diamines. The spacing between helices is measured by X-ray scattering, generating the force-distance curves shown here. As the size of the alkyl chain linking the amines increases, the decay length hydration force seen at low pressures increases. All force curves converge at higher pressures, indicating an insensitivity of the shorter decay length force to linker length. The diamines show a different force behavior from univalent alkyl monoamines. Understanding the origin of those dependencies and differences is critical for linking structure, interaction, and thermodynamics.
Publications
- Lander GC, Johnson JE, Rau DC, Potter CS, Carragher B, Evilevitch A. DNA bending induced phase transition of encapsidated genome in phage lambda. Nucleic Acids Res 2013;41:4518-4524.
- Derouchey JE, Hoover B, Rau DC. A comparison of DNA compaction by arginine and lysine peptides: a physical basis for arginine rich protamines. Biochemistry 2013;52:3000-3009.
- Ferrandino R, Sidorova N, Rau DC. Using single-turnover kinetics with osmotic stress to characterize the EcoRV cleavage reaction. Biochemistry 2014;53:235-246.
- Sae-Ueng U, Li D, Zuo X, Huffman JB, Homa FL, Rau D, Evilevitch A. Solid-to-fluid DNA transition inside HSV-1 capsid close to the temperature of infection. Nat Chem Biol 2014;10:861-867.
- Liu T, Sae-Ueng U, Li D, Lander G, Zuo X, Jonsson B, Rau D, Shefer I, Evilevitch A. Solid-to-fluid DNA transition in viruses facilitates infection. Proc Natl Acad Sci USA 2014;111:14675-14680.
Collaborators
- Jason DeRouchey, PhD, University of Kentucky, Lexington, KY
- Alex Evilevitch, PhD, Carnegie Mellon University, Pittsburgh, PA
- V. Adrian Parsegian, PhD, University of Massachusetts, Amherst, MA
- Xiangyun Qiu, PhD, George Washington University, Washington, DC
Contact
For more information, email raud@mail.nih.gov.