

You are here: Home > Section on Neurophysiology and Biophysics
Glutamate Receptor Structural Biology

- Mark L. Mayer, PhD, Head, Section on Neurophysiology and Biophysics
- Charu Chaudhry, PhD, Postdoctoral Fellow
- Carla Glasser, PhD, Technical Specialist
- Janesh Kumar, PhD, Visiting Fellow
- Jinjin Zhang, PhD, Visiting Fellow
- Anthony Berger, BSc, Postbaccalaureate Fellow
- Suvendu Lomash, PhD, Visiting Fellow
Ionotropic glutamate receptors (iGluRs) are membrane proteins that act as molecular pores and mediate signal transmission at the majority of excitatory synapses in the mammalian nervous system. The seven iGluR gene families in humans encode 18 subunits, which assemble to form three major functional families named after the ligands that were first used to identify iGluR subtypes in the late 1970s: AMPA, kainate, and NMDA. Given the essential role of iGluRs in normal brain function and development, and mounting evidence that dysfunction of iGluR activity mediates several neurological and psychiatric diseases and damage during stroke, we devote substantial effort to analyzing iGluR function at the molecular level. Atomic resolution structures solved by protein crystallization and X-ray diffraction provide a framework for designing electrophysiological and biochemical experiments aimed at defining the mechanisms underlying ligand recognition, the gating of ion channel activity, and the action of allosteric modulators. Information derived from these experiments will permit the development of subtype-selective antagonists and allosteric modulators with novel therapeutic applications and reveal the inner workings of a complicated protein machine that plays a key role in brain function.
Expression and purification of full-length kainate receptor ion channels
Structural studies on CNS membrane proteins are notoriously difficult because commonly used bacterial protein expression systems cannot be used to express eukaryotic membrane proteins. We have recently succeeded in obtaining highly purified preparations of two different kainate receptor subtypes expressed in both insect cells, using baculovirus expression, and HEK 293 cells, using transient transfection. For both systems cultures must be grown on the 12–liter and larger scale in order to obtain sufficient protein for biochemical and structural analysis. Because the amounts of protein obtained are still at least an order of magnitude lower than for soluble proteins, it is necessary to use highly efficient methods for establishing conditions in which the proteins remain, for several days following purification, correctly folded, do not aggregate, or dissociate into dimers and monomers. To do this, we use tryptophan fluorescence size exclusion chromatography (FSEC), for which only microgram quantities of purified protein are required for each run of a 300 mM–length analytical gel filtration column. Using this approach, we have screened numerous detergents, lipids, receptor ligands, and mutants with selective removal of glycosylation sites and various C terminal deletions. Our next goal is to start crystallization trails using conventional detergent–solubilized protein, as well as bicelles and lipidic cubic phases.
Structural studies on the amino-terminal domain of iGluRs
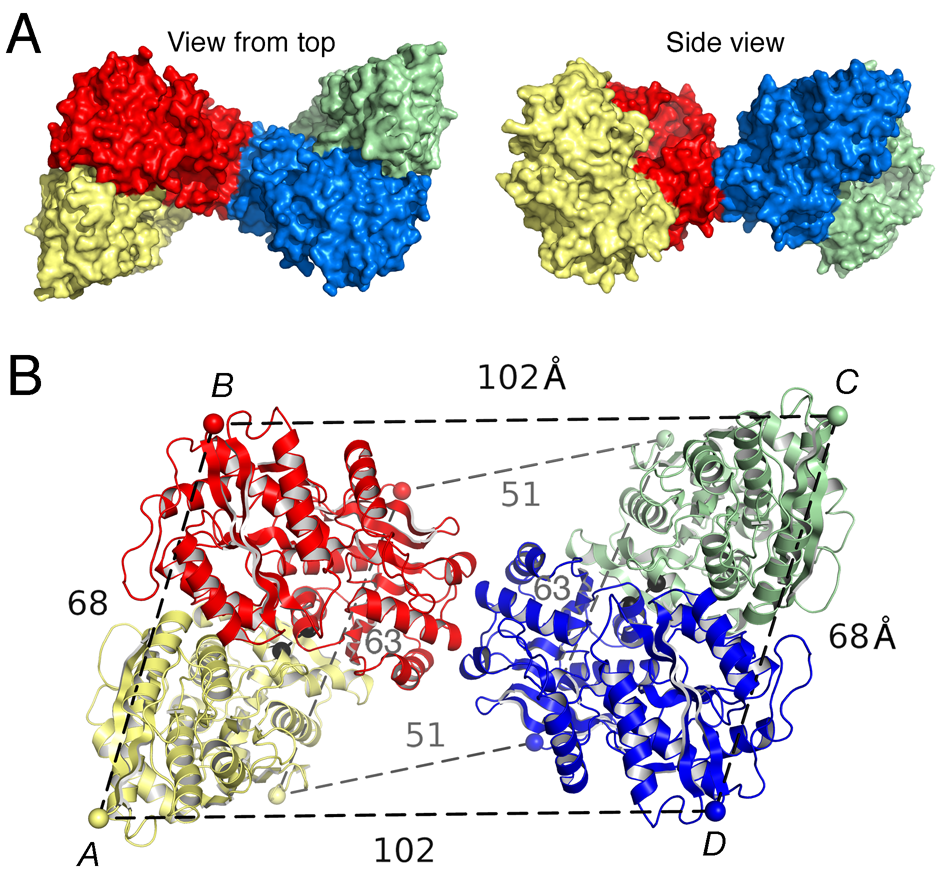
Click image to enlarge.
Figure 1. Crystal structure of the GluK2 (GluR6) ATD tetramer assembly
A. Molecular surface of the crystal structure of the GluR6 (GluK2) ATD tetramer assembly viewed from the top, and, after rotation by 90°, with the 4 subunits colored individually. B. Ribbon diagram showing how the GluK2 ATD AB and CD subunit dimers pack via the lateral edges of domain R2 to form a tetramer; the CA atoms of Thr2 and Glu384, which define the N- and C-termini of the ATD in each subunit, are indicated by colored spheres connected by dashed lines; black dumbbells indicate the CA positions of L151 in the AB and CD dimers.
Glutamate receptor ion channels (iGluRs) are excitatory neurotransmitter receptors with a unique molecular architecture, in which the extracellular domains assemble as a dimer of dimers. The structure of individual dimer assemblies was established previously for both the isolated ligand-binding domain (LBD) and more recently for the larger amino-terminal domain (ATD). How these dimers pack to form tetrameric assemblies in intact iGluRs has remained controversial. Using recently solved crystal structures for the GluK2 kainate receptor ATD as a guide, we performed cysteine mutant cross-linking experiments in full-length tetrameric GluR6 (GluK2) to establish how the ATD packs in a dimer of dimers assembly. A similar approach, using a full-length AMPA receptor GluA2 crystal structure as a guide, was used to design cysteine mutant cross-links for the GluR6 LBD dimer of dimers assembly. The formation of cross-linked tetramers in full-length GluR6 by combinations of ATD and LBD mutants that individually produce only cross-linked dimers suggests that subunits in the ATD and LBD layers swap dimer partners. Functional studies reveal that cross-linking either the ATD or the LBD inhibits activation of GluR6 and that, in the LBD, cross-links within and between dimers have different effects. Our results thus establish that kainate and AMPA receptors have a conserved extracellular architecture and provide insights into the role of individual dimer assemblies in activation of ion channel gating.
In ongoing work, we solved the crystal structure of the GluR7 (GluK3) and KA2 (GluK5) ATDs and made substantial progress in analyzing the mechanisms underlying assembly of heteromeric ATD assemblies, using proteins expressed at the milligram scale in HEK 293 cells. To do this, we solved crystal structures for GluR6/KA2 heterodimer assemblies and for a GluR6/KA2 tetramer assembly. Using these structures as a guide, we performed an extensive analysis of the energetics underling heterodimer assembly, using site-directed mutagenesis to produce mutant proteins. These were purified to homogeneity and then mixed with wild-type proteins, and the Kd for heterodimer assembly was measured by analytical ultracentrifugation (AUC). By examining various combinations, we performed a thermodynamic mutant cycle analysis, which revealed that assembly is an additive process mediated by contacts formed by both the upper and lower lobes of ATD protomers. Electrophysiological experiments revealed that high-affinity contacts mediated by the ATDs of the GluR6 and KA2 subunits are required for formation of heteromeric receptors, even though homomeric receptors can form in ATD− deletion constructs.
Molecular biophysical studies on kainate receptor dimer assembly
For kainate-, but not AMPA-subtype glutamate receptors, the binding of sodium and chloride ions to discrete, electrostatically coupled sites in the extracellular LBD dimer assembly regulates the rate of entry into the desensitized state, which occurs when the dimer interface ruptures and the channel closes. Direct measurement of the effects of allosteric ions on dimer assembly by kainate-subtype iGluRs is not possible with electrophysiological techniques. To obtain proof that allosteric ions regulate dimer formation, we developed a series of GluR6 dimer interface mutations remote from the ion-binding sites and set out to develop a preparation amenable to analysis by AUC. During the course of this project, we encountered issues with buffer mismatch due to the unusual mixture of heavy ions used in our analysis—for example cesium methanesulfonate—and investigated new forms of data analysis to address this.
Sedimentation velocity (SV) analytical ultracentrifugation has re-emerged as an important tool in the characterization of biological macromolecules and nanoparticles. The computational analysis of the evolution of the macromolecular concentration profile allows the characterization of many hydrodynamic and thermodynamic properties of the macromolecules and their interactions. For its usually superior data quality and the wide applicability of refractive index sensitive detection, the Rayleigh interference optical system is often the detection method of choice. However, the interference optical system is also sensitive to the redistribution of co-solvent molecules, which are not of primary experimental interest. In principle, their contribution can be eliminated by an exact geometric and compositional match of the sample solution and the reference solution, achieving the complete optical subtraction of unwanted buffer signals. Unfortunately, in practice, this often cannot be perfectly achieved for various reasons, leading to signal offsets arising from unmatched sedimentation of solvent components. If unrecognized, this can lead to significant misfit, accompanied by significant errors in the macromolecular sedimentation parameters. We developed an approach of computationally accounting for signals from sedimenting buffer components by explicitly modeling their redistribution with Lamm equation solutions, implemented in the software SEDFIT. We demonstrated how this can restore the SV analysis to yield a high-quality fit of the data and to provide correct macromolecular sedimentation parameters.
Crystallographic analysis of kainate receptor competitive antagonists
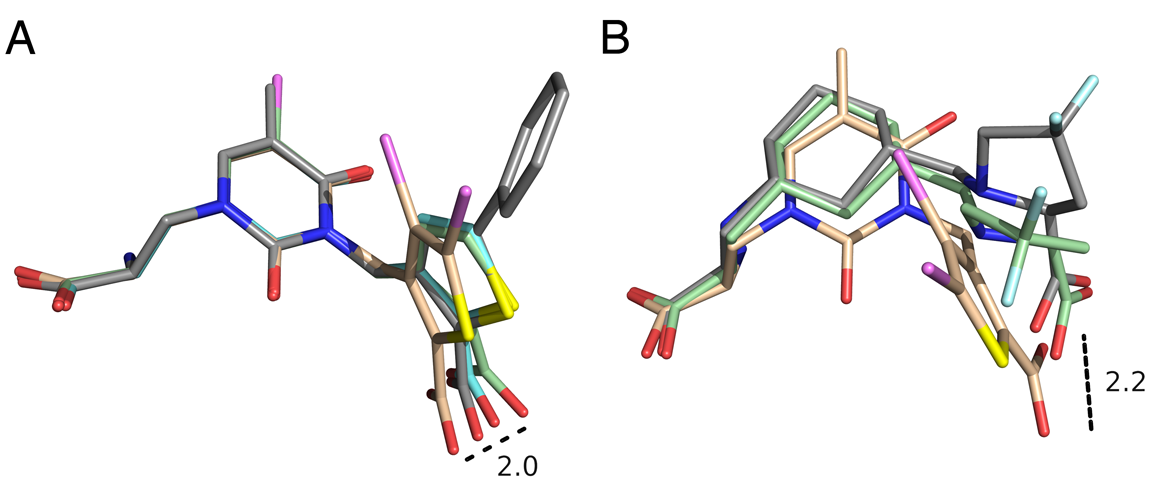
Click image to enlarge.
Figure 2. Conformational variation in the mode of binding of GluK1 (GluR5) competitive antagonists
A. Crystal structures for UBP310, UBP315, UBP316, and UPB 318 with carbon atoms colored cyan, wheat, gray, and green, respectively. The ligand coordinates were extracted from their GluK1 crystal complexes after a prior superimposition using domain 1 protein Ca coordinates, and then rotated so that the uracil rings were superimposed to illustrate how torsion angle variation between the thiophene and uracil rings alters the position of the 2-position carboxyl substituent by up to 2 Å. B. Crystal structures for UBP315 and the up and down conformers of LY466195. The ligand coordinates were extracted from their GluK1 crystal complexes after a prior superimposition using domain 1 protein Ca coordinates, with no further manipulation, and show the large space swept out by the segments of the ligands that interact with the domain 2 pocket located adjacent to the N-terminus of helix F.
The availability of crystal structures for the ligand-binding domains of ionotropic glutamate receptors, combined with their key role in synaptic function in the normal and diseased brain, offers a unique selection of targets for pharmaceutical research, compared with other drug targets for which the atomic structure of the ligand-binding sites is not known. Currently, only a few antagonist structures have been solved, and these reveal ligand-specific conformational changes that hinder rational drug design. We have now solved high-resolution crystal structures for three kainate receptor GluK1 antagonist complexes, which reveal new and unexpected modes of binding, highlighting the continued need for experimentally determined receptor-ligand complexes. The new structures are for GluK1 complexes with (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxy-4,5-dibromothiophene-3-yl-methyl)-5-methylpyrimidine-2,4-dione (UBP315), (S)-1-(2-amino-2-carboxyethyl)-5-bromo-3-(2-carboxythiophene-3-yl-methyl)-pyrimidine-2,4-dione (UBP318), and (3 S,4 aR,6S,8aR)-6-[[(2S )-2-carboxy-4,4-difluoro-1-pyrrolidinyl]-methyl]decahydro-3-isoquinolinecarboxylic acid (LY466195).
The new structures reveal a wide variation in ligand-receptor interactions and ligand binding-domain closure. Given the variation in ligand binding-domain closure, the complexes will be useful for ligand docking and in silico screening of compound libraries. It is interesting that the degree of domain closure is very similar for UBP315 and LY466195, two structurally unrelated ligands, which bind with distinct mechanisms and with high affinity. This suggests that energetic minima do exist in the conformational landscape of large-scale domain rearrangements, yet diverse ligand-binding configurations are compatible with the same domain arrangement. A superposition of the UBP315 and LY466195 ligands in the binding site complex shows just how diverse the ligand binding conformations are. Our understanding of the rules underlying both protein conformational bias and the surprising chemical plasticity of the ligand-binding pocket of iGluRs will benefit from continued crystallographic studies of subtype-selective ligands, as they become available.
Additional Funding
- NICHD IRP
Publications
- Mayer ML. Structure and mechanism of glutamate receptor ion channel assembly, activation and modulation. Curr Opin Neurobiol 2011;21:283-290.
- Kumar J, Schuck P, Mayer ML,. Structure and assembly mechanism for heteromeric kainate receptors. Neuron 2011;71:319-331.
- Das U, Kumar J, Mayer ML, Plested AJ. Domain organization and function in GluK2 subtype kainate receptors. Proc Natl Acad Sci USA 2010;107:8463-8468.
- Kumar J, Mayer ML. Crystal structures of the glutamate receptor ion channel GluK3 and GluK5 amino-terminal domains. EMBO J 2010;404:680-96.
- Kumar J, Schuck P, Jin R, Mayer ML. The N-terminal domain of GluR6 subtype glutamate receptor ion channels. Nat Struct Mol Biol 2009;16:631-638.
Collaborators
- David Jane, PhD, University of Bristol, Bristol, UK
- Peter Schuck, PhD, Protein Biophysics Resource, DBEPS, NIH, Bethesda, MD