

You are here: Home > Section on Nutrient Control of Gene Expression
Transcriptional and Translational Regulatory Mechanisms in Nutrient Control of Gene Expression

- Alan G. Hinnebusch, PhD, Head, Section on Nutrient Control of Gene Expression
- Hongfang Qiu, PhD, Staff Scientist
- Neelam Sen, PhD, Research Fellow
- Sebastien Lagiex, PhD, Postdoctoral Fellow
- Anil Thakur, PhD, Postdoctoral Fellow
- Jyothsna Visweswaraiah, PhD, Postdoctoral Fellow
- Fnu Yashpal, PhD, Postdoctoral Fellow
- David Young, PhD, Postdoctoral Fellow
- Quira Zeiden, PhD, Postdoctoral Fellow
- Fujun Zhou, PhD, Postdoctoral Fellow
- Jinsheng Dong, PhD, Senior Research Assistant
- Fan Zhang, MS, Senior Research Assistant
- Cuihua Hu, BA, Research Assistant
- Laura Marler, BS, Graduate Student
We study molecular mechanisms of gene regulation at the translational and transcriptional levels, using the regulation of amino acid–biosynthetic genes in budding yeast as a model system. Transcription of these and many other genes is coordinately induced by the transcriptional activator Gcn4 in response to starvation of any amino acid. Expression of GCN4 is coupled to amino acid levels by a conserved translational control mechanism involving upstream open reading frames (uORFs) in GCN4 mRNA. Ribosomes translate the 5′-most uORF (uORF1) and, under non-starvation conditions, reinitiate translation at uORFs 2, 3, or 4 and then dissociate from the mRNA, keeping GCN4 translation repressed. In starvation conditions, the reinitiating ribosomes bypass uORFs 2–4 and reinitiate at GCN4 instead, owing to decreased availability of the ternary complex (TC)—comprised of initiation factor 2 (eIF2), GTP, and initiator Met-tRNAi—which binds to the small (40S) ribosomal subunit to assemble a 43S preinitiation complex (PIC). TC abundance is reduced in starved cells by phosphorylation of the alpha subunit of eIF2 (eIF2α) by Gcn2, a protein kinase conserved in all eukaryotes, converting eIF2 from substrate to inhibitor of its guanine nucleotide exchange factor (GEF) eIF2B. Hence, GCN4 translation is an in vivo indicator of impaired TC loading on 40S subunits. We previously exploited this fact to isolate mutations in subunits of eIF2B that constitutively derepress GCN4 (Gcd− phenotype) by lowering TC assembly in the absence of eIF2 phosphorylation. More recently, we used the Gcd− selection to identify domains/residues in eIF1, eIF1A, and eIF3, and also residues of 18S rRNA located near the “P” decoding site of the 40S subunit, that participate in rapid TC recruitment in vivo. In collaboration with Jon Lorsch's group, we demonstrated that segments/residues in eIF1, eIF1A, and 18S rRNA, which are implicated genetically in TC recruitment, also stimulate this reaction in a fully reconstituted in vitro system.
We also investigate the roles of various eIFs and the 40S subunit in scanning the mRNA 5′ untranslated region and accurately identifying the AUG initiation codon. The studies exploit a genetic selection for mutations that elevate initiation at near-cognate UUG start codons (Sui− phenotype) or suppress this aberrant initiation event (Ssu− phenotype). In this way, we showed that eIF1A and the c-subunit of eIF3 make critical contributions to accurate AUG recognition, and localized these functions to the unstructured N- and C-tails of eIF1A and the N-terminal domain of eIF3c. Also in collaboration with Lorsch's group, we provided strong evidence that dissociation of eIF1 from the PIC is a critical step in AUG recognition, indicating that eIF1 serves as a "gate-keeper" necessary to suppress initiation at non-AUG triplets. Biochemical analysis revealed that eIF1 stabilizes an "open" conformation of the 40S subunit conducive to scanning and loading of TC in a metastable state (POUT), which enables inspection of successive mRNA triplets in the P site for complementarity with the anticodon of Met-tRNAi. However, eIF1 must dissociate from the PIC for AUG selection, as the absence of eIF1 favors the closed, scanning-arrested conformation of the 40S subunit with TC more tightly bound (PIN state). We recently established that basic residues in eIF1's helix-1 and β-hairpin-1 mediate 40S binding and stabilize the open, POUT complex, to promote Met-tRNAi recruitment and accurate AUG selection in vivo. Previously, we showed that 10–amino acid repeats in the C-terminal tail (CTT) of eIF1A (dubbed "scanning enhancers", or SE elements) function together with eIF1 to stabilize the open, POUT complex but must be antagonized by scanning-inhibitory (SI) elements in the N-terminal tail (NTT) and helical domain of eIF1A to achieve rearrangement to the closed, PIN complex and start codon recognition. In collaboration with Lorsch's group, we recently established that the eIF1A CTT moves towards the N-terminal domain (NTD) of eIF5 in response to AUG recognition, and that this movement—which depends on the eIF1A SE elements—is required for Pi release from eIF2. We proposed that accommodation of tRNAi in the PIN state triggers eIF1 displacement, which in turn triggers movement of the eIF1A-CTT and Pi release.
The eIF4F complex, which binds to the mRNA 5′ cap structure, is thought to resolve mRNA secondary structure to enhance recruitment of the 43S PIC and scanning of the mRNA leader. We recently dissected the NTD of eIF4G, the scaffolding subunit of eIF4F—harboring binding sites for cap-binding factor eIF4E, poly(A)-tail-binding factor PABP, and RNA helicase eIF4A—and identified conserved elements (RNA1, boxes 1–3) that functionally overlap to promote eIF4G interaction with mRNA and PABP, thereby activating mRNA for PIC attachment. We also identified a novel role for cofactor eIF4B in promoting eIF4AVeIF4G association (eIF4F assembly) in vivo; and in collaboration with Lorsch's group, showed that binding of eIF4B near the mRNA entry channel of the 40S subunit is likely to be crucial for stimulating eIF4F function in PIC recruitment.
In the arena of transcriptional control, we previously defined multiple clusters of hydrophobic residues that constitute the transcriptional activation domain of Gcn4, identified co-activators required for gene activation by Gcn4 in vivo, and defined the molecular program for recruitment of nucleosome-modifying and -remodeling enzymes and adaptor proteins that remove repressive chromatin structure and recruit TATA–binding protein and RNA polymerase II (Pol II) to Gcn4 target promoters. We also demonstrated co-transcriptional recruitment of the histone acetyltransferase (HAT) complexes SAGA and NuA4 to transcribed coding sequences and showed that they cooperate to enhance transcription-associated histone eviction and Pol II elongation in vivo. We described a two-stage recruitment mechanism for NuA4, involving the serine-5–phosphorylated C-terminal domain (CTD) of Pol II and methylated histones, and more recently we extended this mechanism to include the histone deacetylase (HDA) complex Rpd3C(S). In addition, we showed that the HDAs Rpd3, Hos2, and Hda1 have overlapping functions in deacetylating coding-sequence nucleosomes and obtained evidence that histone acetylation is a key determinant of co-transcriptional nucleosome disassembly.
More recently, we elucidated the roles of the Pol II CTD kinases Kin28 and Bur1 and the C-terminal repeats (CTRs) of the Spt5 subunit of DSIF in recruitment of transcription elongation factor Paf1C. We showed that Ser5-CTD phosphorylation by Kin28 enhances recruitment of Bur1 near promoter regions where Bur1 contributes to Ser2–CTD phosphorylation. We then established that Kin28 enhances Paf1C recruitment by (i) promoting Bur1 recruitment via the phosphorylated Pol II CTD, with attendant phosphorylation of Spt5 CTRs by Bur1; and (ii) collaborating with Bur1 to generate Ser2–/Ser5–diphosphorylated Pol II CTD repeats. The resulting phosphorylated CTD and CTR repeats bind to distinct Paf1C subunits to enable efficient recruitment of the entire complex to elongating Pol II. We also showed that disrupting vesicular trafficking at the endosome attenuates transcriptional activation by Gcn4, and, in collaboration with Vytas Bankaitis, implicated the sterol-binding protein Kes1, sphingolipid signaling to the nucleus, and Cdk8/Srb10 in mediating this response. We further implicated various proteins with established functions in vesicular protein sorting (Vps), particularly the PI 3-kinase Vps34 and the serine/threonine protein kinaseVps15, in gene gating at nuclear pores and in transcription elongation in the nucleus.
Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy.
Recognition of the start codon is thought to require dissociation of eIF1 from the 40S subunit, enabling Pi release by the TC, rearrangement of the 40S subunit to a closed conformation incompatible with scanning, and stable binding of Met-tRNAi to the P site. Supporting this model, we showed previously that eIF1 mutations that impair its binding to 40S subunits in vitro increase initiation at UUG codons (Sui− phenotype) and derepress translation of GCN4 mRNA (Gcd− phenotype), indicating impaired TC loading. This indicated that direct contact of eIF1 with 18S rRNA is crucial for eIF1's ability to stabilize the open conformation of the 40S subunit competent for rapid loading of TC in a conformation capable of sampling triplets entering the P site (“POUT”), while blocking accommodation of tRNAi in the “PIN” state required for AUG recognition. Recently, we selected eIF1 Ssu− mutations that suppress the elevated UUG initiation and reduced rate of TC loading in vivo conferred by aforementioned eIF1 Sui− substitutions. Importantly, several such Ssu− substitutions increase eIF1 affinity for 40S subunits in vitro; the strongest binding variant (D61G), predicted to eliminate ionic repulsion with 18S rRNA, both reduces the rate of eIF1 dissociation and destabilizes the PIN state of TC binding in PICs reconstituted with Sui− variants of eIF5 or eIF2. Thus, eIF1 dissociation from the 40S subunit is required for the PIN mode of TC binding and AUG recognition, and augmenting eIF1's affinity for the 40S subunit increases initiation accuracy in vivo.
Eukaryotic translation initiation factor eIF5 promotes the accuracy of start codon recognition by regulating Pi release and conformational transitions of the preinitiation complex.
Previous work suggested that the eIF5 Sui− mutation G31R/SUI5 elevates UUG initiation by increasing eIF5 GAP function. Subsequent work implicated eIF5 in rearrangement of the PIC from the open to closed conformation at AUG codons, from which Pi is released. To identify eIF5 functions crucial for accurate initiation, we investigated the consequences of G31R on GTP hydrolysis and Pi release, and the effects of intragenic G31R suppressors on these reactions and on the partitioning of PICs between the open and closed states. eIF5-G31R altered regulation of Pi release, accelerating it at UUG while reducing it at AUG codons, consistent with its known ability to stabilize the closed PIC conformation at UUG codons (as manifested by tighter binding of eIF1A to the PIC). Suppressor G62S mitigates both consequences of G31R, accounting for its efficient suppression of UUG initiation in G31R,G62S cells; however, suppressor M18V impairs GTP hydrolysis but has little effect on PIC conformation. The strong defect in GTP hydrolysis conferred by M18V likely explains its broad suppression of Sui− mutations in numerous factors. We conclude that both of eIF5's functions, regulating Pi release and stabilizing the closed PIC conformation, contribute to stringent AUG selection in vivo, and our results identify different segments of eIF5 involved in regulating these distinct functions.
Conserved residues in yeast initiator tRNA calibrate initiation accuracy by regulating preinitiation complex stability at the start codon.
Eukaryotic tRNAi contains unique sequence features, but their importance in start codon selection was unknown. We found that disrupting the conserved C3:G70 base pair in the acceptor stem enhances initiation at UUG codons (Sui− phenotype) and also reduces the rate of TC binding to 40S subunits both in vitro and in vivo (Gcd− phenotype). The defects are suppressed by an Ssu− substitution in the eIF1A SI element shown previously to stabilize the open/POUT conformation. Because the consequences of C3:G70 substitutions mimic the known effects of Sui− mutations in eIF1 and eIF1A SE elements, it appears that the C3:G70 base pair functionally interacts with eIF1 and eIF1A to promote the POUT conformation, favoring rapid TC binding and impeding rearrangement to the PIN conformation at non-AUG codons. Substituting the conserved G31:C39 base pair in the anticodon stem with various base pairs also reduces initiation accuracy (Sui− phenotype) but does not impair TC binding to the PIC, suggesting that base-pair replacements at 31:39 enhance UUG initiation by stabilizing the PIN state at near-cognate triplets rather than destabilizing the POUT conformation. By contrast, mutations that eliminate base pairing at 31:39 have the opposite effect of increasing initiation accuracy (Ssu− phenotype), which is suppressed by a Sui− substitution of T-loop residue A54 in tRNAi. These opposing genetic phenotypes are paralleled by opposite effects of the Sui− and Ssu− substitutions on the stability of Met-tRNAi binding to reconstituted PICs in vitro. We propose that any of the three alternative Watson-Crick base pairs at position 31:39 is sufficient to stabilize PIN and promote initiation by inherently less stable PICs formed at UUG codons. However, the (wild-type) G31:C39 pair uniquely imposes an energetic penalty on the PIN state that is compensated effectively only with the perfect codon:anticodon duplex at AUG codons. Thus, conserved bases throughout tRNAi, from the anticodon stem to acceptor stem, play key roles in ensuring the efficiency and fidelity of start codon recognition in vivo (summarized in Figure 1).
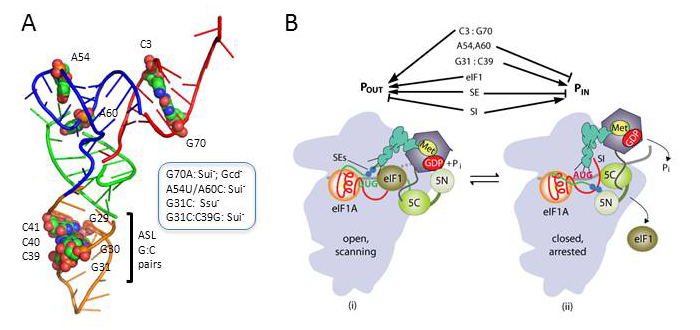
Click image to enlarge.
Figure 1. Conserved residues in yeast initiator tRNA calibrate initiation accuracy by regulating preinitiation complex stability at the start codon.
(A) PyMol rendering of the crystal structure of yeast tRNAi (PDB: 1YFG) using color-coding to designate the acceptor stem (red), T-stem–loop (green), D-stem–loop (blue), and ASL (gold) and depicting, by spheres, bases or base pairs we implicated in start codon recognition. Phenotypes of substitutions disrupting the key anticodon stem base pair G31:C39 (G31C, Ssu− [hyperaccuracy]) or replacing it with base pair C31:G39 (Sui−, [hypoaccuracy]), substituting T-loop residues A54/A60 (Sui−) or disrupting C3:G70 in the acceptor stem (G70A, Sui− and Gcd− [slow TC loading]) are summarized in the box. (B) Model summarizing the deduced roles of conserved tRNAi residues in start codon recognition. The open/POUT and closed/PIN states of the PIC and roles of eIF1 and the SE/SI elements of eIF1A in regulating conformational rearrangements and reactions accompanying AUG recognition are depicted (see text). C3:G70 functions together with eIF1 and eIF1A to stabilize the POUT conformation of TC binding, whereas A54/A60 impede rearrangement to the PIN state in a manner overcome efficiently only with the perfect codon:anticodon duplex formed at AUG. G31:C39 is required for thermodynamic coupling between AUG and tRNAi in the PIN state, and, with other base pairs (e.g., C31:G39), it further stabilizes PIN to increase initiation at NUG near-cognate codons.
Identification and characterization of functionally critical, conserved motifs in the internal repeats and N-terminal domain of yeast translation initiation factor 4B (yeIF4B)
The translation initiation factor eIF4B stimulates recruitment of mRNA to the 43S PIC, but its molecular function is unclear. The yeast (y)eIF4B contains an unstructured N-terminal domain (NTD), RNA–recognition motif (RRM), and a domain comprising seven imperfect repeats of 26 amino acids; previous studies had implicated the RRM and its RNA binding activity in promoting translation initiation. By analyzing the effects of deletions and mutations of yeIF4B domains on PIC attachment to mRNA in vitro and translation initiation in vivo, we found that the 7-repeat domain is critical for productive interaction with the PIC and other components of the initiation machinery, particularly eIF4A, in promoting PIC attachment to mRNA. The NTD also plays a role in accelerating mRNA binding to the PIC but, surprisingly, the RRM and its associated ssRNA–binding activity are dispensable in vitro and in vivo. We recently determined that only two of the seven internal repeats are sufficient for wild-type (WT) yeIF4B function in vivo when all other domains are intact, whereas at least three are needed in the absence of the NTD or when functions of eIF4F components are compromised. We corroborated these observations by demonstrating that yeIF4B variants with only one or two repeats display substantial activity in promoting mRNA recruitment by the PIC in vitro, whereas additional repeats are required at lower levels of eIF4A or when the NTD is missing. We also demonstrated that only three highly conserved positions in the 26-aa repeat are essential for function in vitro and in vivo, identified conserved motifs in the NTD, and demonstrated functional overlap of two such motifs. The results provide a comprehensive description of the critical sequence elements in yeIF4B that support eIF4F function in mRNA recruitment by the PIC (Zhou F, Walker SE, Mitchell SF, Lorsch JR, Hinnebusch AG. Characterization of functionally critical, conserved motifs in the internal repeats and N-terminal domain of yeast translation initiation factor 4B (yeIF4B). J Biol Chem 2014;289:1704-1722).
Enhanced interaction between pseudokinase and kinase domains in Gcn2 stimulates eIF2α phosphorylation in starved cells.
The stress-activated protein kinase Gcn2 regulates protein synthesis by phosphorylating the α subunit of eIF2, from yeast to mammals. The Gcn2 kinase domain (KD) is inherently inactive and requires allosteric stimulation by adjoining regulatory domains. Gcn2 contains a pseudokinase domain (YKD) required for high-level eIF2α phosphorylation in amino acid–starved yeast cells; however, the role of the YKD in KD activation was unknown. We isolated substitutions of evolutionarily conserved YKD amino acids that impair Gcn2 activation without reducing binding of the activating ligand, uncharged tRNA, to the histidyl-tRNA synthetase–related domain of Gcn2. Several such Gcn− substitutions cluster in predicted helices E and I (αE and αI) of the YKD. We also identified Gcd− substitutions, evoking constitutive activation of Gcn2, mapping in αI of the YKD. Interestingly, αI Gcd− substitutions enhance YKD–KD interactions in vitro, whereas Gcn− substitutions in αE and αI suppress both this effect and the constitutive activation of Gcn2 conferred by YKD Gcd− substitutions. The findings indicate that the YKD interacts directly with the KD for activation of kinase function and identify likely sites of direct YKD–KD contact. We propose that tRNA binding to the HisRS domain evokes a conformational change that increases access of the YKD to sites of allosteric activation in the adjoining KD.
Accumulation of a threonine-biosynthetic intermediate attenuates general amino acid control by accelerating degradation of Gcn4 via the Cdks Pho85 and Cdk8/Srb10.
Gcn4 abundance is tightly regulated by the interplay between the intricate translational control mechanism that induces Gcn4 synthesis in starved cells and a pathway of phosphorylation and ubiquitylation that mediates its rapid degradation by the proteasome, involving the cyclin-dependent kinases (Cdks) Pho85 and Srb10/Cdk8. We discovered that transcriptional activation by Gcn4 is attenuated in mutants lacking the threonine biosynthetic enzyme encoded by HOM6, and determined that accumulation of the Hom6 substrate beta-aspartate semialdehyde (ASA) is responsible for the reduced activation of Gcn4 target genes in hom6Δ cells. Remarkably, ASA accumulation was found to accelerate the already rapid degradation of Gcn4 in a manner requiring its phosphorylation by Srb10 and Pho85. Our analysis of Gcn4 promoter occupancy and activation function of Gcn4 molecules rescued from degradation in srb10Δ versus pho85Δ cells unveiled an unexpected division of labor between the two Cdks. Whereas Srb10 primarily targets inactive Gcn4 molecules—presumably damaged under conditions of ASA excess—Pho85 clears a greater proportion of functional Gcn4 species from the cell. The ability of ASA to inhibit transcriptional induction of threonine-pathway enzymes by Gcn4, dampening ASA accumulation and its toxic effects on other aspects cell physiology, is expected to be adaptive in the wild when yeast encounters natural antibiotics that target Hom6 enzymatic activity.
Additional Funding
- International Human Frontiers of Science Program Grant RGP0028
Publications
- Martin-Marcos P, Nanda JS, Luna RE, Zhang F, Saini AK, Cherkasova VA, Wagner G, Lorsch JR, Hinnebusch AG. Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy. RNA 2014;20:150-167.
- Saini AK, Nanda JS, Martin-Marcos P, Dong J, Zhang F, Bhardwaj M, Lorsch JR, Hinnebusch AG. Eukaryotic translation initiation factor eIF5 promotes the accuracy of start codon recognition by regulating Pi release and conformational transitions of the preinitiation complex. Nucleic Acids Res 2014;42:9623-9640.
- Dong J, Munoz A, Kolitz SE, Saini AK, Chiu WL, Rahman H, Lorsch JR, Hinnebusch AG. Conserved residues in yeast initiator tRNA calibrate initiation accuracy by regulating preinitiation complex stability at the start codon. Genes Dev 2014;28:502-520.
- Lageix S, Rothenburg S, Dever TE, Hinnebusch AG. Enhanced interaction between pseudokinase and kinase domains in Gcn2 stimulates eIF2α phosphorylation in starved cells. PLoS Genet 2014;10:e1004326.
- Rawal Y, Qiu H, Hinnebusch AG. Accumulation of a threonine biosynthetic intermediate attenuates general amino acid control by accelerating degradation of Gcn4 via Pho85 and Cdk8. PLoS Genet 2014;10:e1004534.
Collaborators
- Katsura Asano, PhD, Kansas State University, Manhattan, KS
- Vytas A. Bankaitis, PhD, Texas A&M University Health Science Center, College Station, TX
- Rachel Green, PhD, The Johns Hopkins University School of Medicine, Baltimore, MD
- Nicholas Ingolia, PhD, University of California Berkeley, Berkeley, CA
- Anton Komar, PhD, Cleveland State University, Cleveland, OH
- Jon Lorsch, PhD, The Johns Hopkins University School of Medicine, Baltimore, MD
- Venkatraman Ramakrishnan, PhD, MRC Laboratory of Molecular Biology, Cambridge, UK
- Gerhard Wagner, PhD, Harvard Medical School, Boston, MA
- Chi-Ming Wong, PhD, University of Hong Kong, Hong Kong, China
Contact
For more information, email hinnebua@mail.nih.gov or visit sncge.nichd.nih.gov.