

You are here: Home > Section on Eukaryotic Transposable Elements
The Biological Impact and Function of Transposable Elements

- Henry L. Levin, PhD, Head, Section on Eukaryotic Transposable Elements
- Angela Atwood-Moore, BA, Senior Research Assistant
- Caroline Esnault, PhD, Visiting Fellow
- Sudhir Rai, PhD, Visiting Fellow
- Parmit Singh, PhD, Visiting Fellow
- Anthony J. Hickey, PhD, Postdoctoral Fellow
- Si Young Lee, PhD, Postdoctoral Fellow
- Maya Sangesland, BA, Postbaccalaureate Fellow
- Georges-Ibrahim A C Cisse, Summer Student
- Shannon Jin, Summer Student
Inherently mutagenic, the integration of retroviral and retrotransposon DNA is responsible for many pathologies, including malignancy. Given that some chromosomal regions are virtually gene free while others encode genes essential for cellular processes, the position of integration has great significance. Recent studies showed clearly that integration occurs into specific types of sequences and that the targeting patterns vary depending on the retrovirus or retrotransposon. Currently, there is great interest in such patterns, in part because understanding the mechanisms that position HIV-1 insertions may lead to new antiviral therapies. In addition, retrovirus-based vectors are now being used for gene therapy. Early gene therapy vectors had patterns of integration that activated oncogenes and caused leukemia in patients. Therefore, to gauge the risks associated with new gene therapy vectors, it is essential that we characterize in detail the positions of integration and understand the mechanisms that position such integration.
Ultra-high throughput sequencing of transposon integration with serial number technology provides a saturated profile of target activity in Schizosaccharomyces pombe.
The finding that integration in the genome of S. pombe is directed to the promoters of genes raises several important questions about the biology of Tf1 integration, namely, whether all promoters are recognized equally or whether integration is directed to specific sets of promoters. If specific sets of promoters are preferred targets, what distinguishes the preferred promoters from those not recognized by Tf1? To address this question, we sequenced large numbers of integrations throughout the genome of S. pombe (Majumdar et al., J Virol 2011;85:519). The revolutionary new methods for ultra-high throughput sequencing made it possible to characterize extraordinarily large numbers of integration events.
To select for the cells with integration events, we induced cells for the expression of Tf1 containing the neo gene (Tf1–neo). We applied ligation-mediated PCR to generate libraries of Tf1–neo associated with the downstream flanking DNA. The amplified products, consisting of the downstream long-terminal repeats (LTRs) and their flanking DNA, were size-selected and submitted for sequencing.
Over 90% of the insertions occurred within intergenic sequences that contained promoters. The profile of integration into the promoter regions revealed substantial variation. We observed reproducibly high levels of integration in 20% of the intergenic sequences in S. pombe. The strong bias is a consequence of the integration preference for a specific set of promoters. We found that there was no correlation between the promoters with the highest transcription activity and the promoters that had high levels of integration. However, the results of a gene ontology analysis revealed that genes regulated by environmental stress are preferred targets of integration.
The size and number of the integration experiments resulted in reproducible measures of integration for each intergenic region and ORF (open reading frame) in the S. pombe genome. However, to understand which factors could mediate integration within these promoters, we needed to know not only where the insertions occurred but how often integration occurred at each nucleotide position, which, to date, we have not been able to determine, because independent integration events at the same nucleotide would result in duplicate sequence reads that would be indistinguishable from the duplicates generated during the PCR amplification of the library. This year, we developed a technique that can measure the number of independent integration events that occur at single nucleotide positions. The technique, termed the serial number system, is based on randomizing eight base pairs in the tip of the Tf1 transposon. Each independent integration event is tagged with the "serial number" of the individual Tf1 element that was inserted. As a result, we can now record as many as 65,000 independent insertions at each nucleotide of the S. pombe genome. Our first application of the technique detected 1.2 million independent insertions and created a saturated and reproducible measure of integration at each nucleotide of S. pombe. To identify factors that may mediate integration, the data are now being compared with the binding sites of transcription factors. Use of the serial number system can be generalized, and we are currently testing it to measure integration levels of retroviruses.
Transposon integration increases the expression of stress-response genes.
Transcription of transposons can be activated when cells are under stress, and environmental stress has been shown to induce integration events. The Nobelist Barbara McClintock put forward the intriguing hypothesis that transposon insertions triggered by conditions of stress may benefit the host by improving survival. However, this model remains unsubstantiated.
The LTR–retrotransposon Tf1 of S. pombe integrates into the promoters of pol II–transcribed genes. Saturated profiles of insertion sites revealed that Tf1 integrates with a preference for pol II promoters that are induced by environmental stresses (Majumdar et al., J Virol 2011;85:519). To determine the biological impact of integration, we examined the effect of Tf1 integration on the expression of the adjacent genes (2). We studied 32 genes often targeted by Tf1 and found that integration did not reduce their expression. In six cases, Tf1 insertion actually increased the expression of adjacent genes by enhancing the levels of the native transcripts. In other cases, host factors that participate in genome surveillance, such as Upf1 and Abp1, were found to restrict the expression of genes that would otherwise have been enhanced by Tf1 insertion. We found that Tf1 transcription was induced by heat treatment and, interestingly, only genes that themselves were induced by heat could be activated by Tf1 integration. We propose that it is the synergy between Tf1 enhancer sequence and the stress-response elements of target promoters that results in gene activation. In support of this model, the motif identification software MEME identified a sequence that was present in the promoter of Tf1 and in those of the six genes enhanced by Tf1 insertion. Importantly, the motif was not present in the 26 promoters that were unaffected by Tf1 insertion. Moreover, the motif is similar to the sequence known to be bound by Atf1, a stress-response transcription factor. Together, the findings indicate that Tf1 inserts can increase the expression of stress-response genes because Tf1 carries a copy of an enhancer that binds to the same factor or factors that stimulate the stress-response genes. We therefore speculate that Tf1 integration has the potential to improve the survival of individual cells exposed to environmental stress.
Tf1 integration improves resistance to environmental stress.
The integration of Tf1 into stress-response promoters together with its ability to increase the expression of these genes suggests that Tf1 may benefit cells exposed to stress by promoting adaptation, consistent with our recent observation that Tf1 transcription and transposition are induced by environmental stress. To test whether Tf1 insertions provide benefit to cells exposed to environmental stress, we grew several cultures containing approximately 50,000 insertions for 80 generations in restrictive concentrations of CoCl2. Deep sequencing of integration sites revealed a reproducible profile of insertion sites that are heavily enriched in cells grown in CoCl2. The enriched insertions were positioned next to 17 specific genes that have functions consistent with tolerance to heavy metals. The results indicate that Tf1 insertion at a variety of sites raises resistance to CoCl2. Three Tf1 insertions that were highly enriched during growth in CoCl2 were recreated in cells that were not exposed to CoCl2. Each of these insertions resulted in resistance to CoCl2, and the resistance was attributed to changes in expression of genes adjacent to Tf1. Some of the genes adjacent to the enriched insertions were associated with the TORC2 pathway of stress response factors. We are currently testing whether TORC2 plays a role in mitigating the toxicity of CoCl2.
Integration profiling: a whole-genome analysis of sequence function
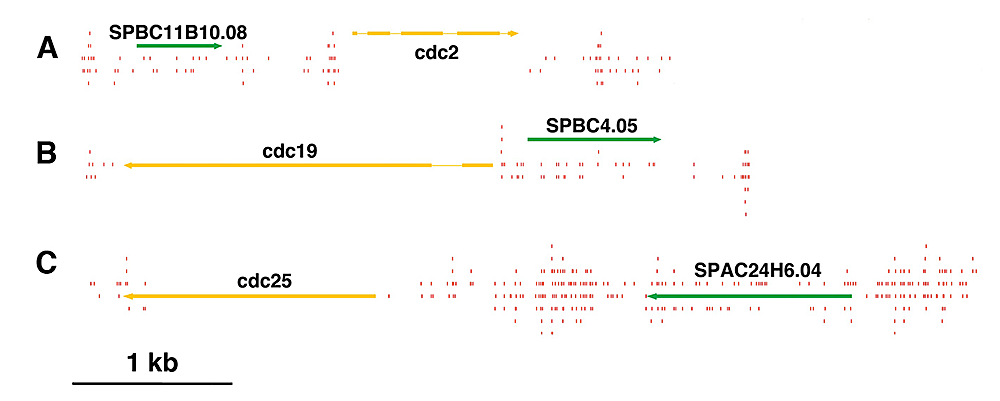
Click image to enlarge.
Figure 1. Essential genes contain low numbers of integration events.
Few insertions (red lines) occurred in essential genes such as the cdc genes (yellow) compared with nonessential genes (green).
The existing genome-wide methods for testing gene function consist largely of microarray hybridization and deep sequencing of RNA, techniques that infer function from patterns of gene expression. Despite the valuable information produced by these methods, they do not provide a direct demonstration of gene function. To address this need, we developed integration profiling, a simple method capable of directly probing the function of the single-copy sequences throughout the genome of a haploid eukaryote. With transposons that readily disrupt ORFs and sequencing technology that can position over 250 million insertions per reaction, the analysis of a single culture can identify which sequences in a eukaryotic genome are functional. In previous work, we found that the “cut and paste” DNA transposon Hermes from the housefly is highly active in S. pombe. The high rate of integration and the disruption of ORFs mean that Hermes is suitable for mutagenesis studies. With integration profiling, large populations of cells with transposon insertions are grown for many generations, depleting the culture of cells that have insertions in genes important for division. In one experiment, we passaged cells for 74 generations until 13.4% of the cells in the final culture contained an integrated copy of Hermes. We determined the positions of the insertions in the culture by ligation-mediated PCR followed by Illumina sequencing. We identified 360,000 unique insertion events that produced an average of one insertion for every 29 bp of the S. pombe genome. A survey of known essential genes revealed very few insertions per ORF, whereas neighboring nonessential gene ORFs had high numbers of insertions (Figure 1). Recently, a consortium systematically deleted the ORFs of S. pombe in heterozygous diploids and, after sporulation, designated which ORFs were essential (Kim et al., Nat Biotechnol 2010;28:617). Using these designations, we plotted the distribution of integration densities separately for the nonessential and essential ORFs. We also graphed the integration densities of a subclass of nonessential genes that, when deleted, resulted in small colonies. Clearly, the essential ORFs had significantly fewer insertions/kb than the nonessential ORFs, indicating that the integration profiles did indeed discriminate between essential and nonessential ORFs. Importantly, the nonessential ORFs required for full colony growth had intermediate densities of integration, indicating that intermediate levels of integration may be used to identify nonessential genes that nevertheless contribute to growth. The principal discrepancy between the designations made by the consortium and the Hermes integration is the group of 200 ORFs designated nonessential, which exhibited very low levels of integration. Using PCR and DNA blotting, we found that the majority of these consortium designations were incorrect because the genes had not been successfully deleted. The results validate integration profiling as an accurate method for measuring gene function.
We extended the use of integration profiling to identify genes important for the formation of heterochromatin. Our initial strain contained a copy of ura4 within the centromeric sequence. The heterochromatin present in the centromeric sequence silenced the expression of ura4 and, as a result, allowed cells to grow in the presence of 5-fluorooritic acid (FOA). We then induced Hermes transposition and passaged cultures for many generations. Disruption of genes required for heterochromatin allowed ura4 to be expressed and, as a result, inhibited growth in a medium containing FOA. To identify the positions that tolerated disruption, we sequenced the integration sites of cells in the final culture. Our data set of one million integration positions contained, on average, one insertion for every 8 bp of the genome. We found that approximately 200 genes contained significantly fewer insertions than the remainder of the genome. Importantly, this gene set contained the majority of genes previously shown to contribute to heterochromatin formation. To test directly their contribution to heterochromatin and to characterize their mode of action, we are now analyzing candidates identified by integration profiling that have not previously been studied.
Analysis of one million independent HIV-1 integration sites identifies a link with mRNA splicing.
The interaction of HIV-1 integrase and the host chromatin-binding factor LEDGF preferentially targets HIV-1 integration to actively transcribed genes. However, it is not clear how LEDGF is recruited to the active genes. Moreover, there is little information showing whether HIV integration favors specific types of genes. To identify chromatin features and cellular factors that play an important role in integration of HIV, we created a map of three million independent HIV insertions in cultured cells. We developed a method that measured independent insertions at single-nucleotide positions and used the method with ligation-mediated PCR and Illumina sequencing. We obtained highly reproducible integration densities per gene in independent PCR libraries and different cell lines. The ontology of the 1,000 genes that were the most favored for integration revealed a substantial enrichment of nuclear mRNA splicing factors, histone methyltransferases, and proteins containing RRM (RNA recognition motif) RNA–binding domains. Oncogenes were approximately five times more common than expected based on their abundance in the genome. Such integration site preference suggests that HIV-1 could impact the biology of infected cells. Integration sites were significantly fewer in regions with low nucleosome occupancy, for example, at promoters and at the first splice junctions, supporting a role of nucleosomes in integration. Integration in intron-less genes favors the 3′ end of the genes, and integration in intron-containing genes favors the 5′ ends of the genes. We observed that integration density in genes correlated strongly with the number of introns in the gene, and RNA-Seq analysis showed that integration correlated with the number of alternative isoforms of the gene, suggesting a link between splicing and LEDGF–dependent HIV-1 integration. The correlation is not dependent on the rate of transcription or length of introns. In genes that are expressed at low levels, there were approximately equal numbers of integration sites throughout the gene, suggesting a role of transcription in the distribution of integration sites. Thus, our analysis of one million integration events indicates that LEDGF–dependent HIV-1 integration involves an interaction with nucleosomes and splicing machinery. In addition, the patterns of integration observed provide insight into the distribution of LEDGF across transcription units and the role of LEDGF in transcription.
Additional Funding
- NIH Intramural AIDS Targeted Antiviral Program (2013 and 2014)
Publications
- Levin H, Moran J. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 2011;12:615-627.
- Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, Wapinski I, Roy S, Lin MF, Heiman DI, Young SK, Furuya K, Guo Y, Pidoux A, Chen HM, Robbertse B, Goldberg JM, Aoki K, Bayne EH, Berlin AM, Desjardins CA, Dobbs E, Dukaj L, Fan L, FitzGerald MG, French C, Gujja S, Hansen K, Keifenheim D, Levin JZ, Mosher RA, Müller CA, Pfiffner J, Priest M, Russ C, Smialowska A, Swoboda P, Sykes SM, Vaughn M, Vengrova S, Yoder R, Zeng Q, Allshire R, Baulcombe D, Birren BW, Brown W, Ekwall K, Kellis M, Leatherwood J, Levin H, Margalit H, Martienssen R, Nieduszynski CA, Spatafora JW, Friedman N, Dalgaard JZ, Baumann P, Niki H, Regev A, Nusbaum C. Comparative functional genomics of the fission yeasts. Science 2011;332:930-936.
- Guo Y, Park JM, Cui B, Humes E, Gangadharan S, Hung S, Fitzgerald PC, Hoe KL, Grewal SI, Craig NL, Levin HL. Integration profiling of gene function with dense maps of transposon integration. Genetics 2013;195:599-609.
- Feng G, Leem YE, Levin HL. Transposon integration enhances expression of stress response genes. Nucleic Acids Res 2013;41:775-789.
- Chatterjee AG, Esnault C, Guo Y, Hung S, McQueen PG, Levin HL. Serial number tagging reveals a prominent sequence preference of retrotransposon integration. Nucleic Acids Res 2014;42:8449-8460.
Collaborators
- Nancy Craig, PhD, The Johns Hopkins Medical School, Baltimore, MD
- Shiv Grewel, PhD, Laboratory of Biochemistry and Molecular Biology, NCI, Bethesda, MD
- Stephen Hughes, PhD, Retroviral Replication Laboratory, HIV Drug Resistance Program, NCI, Frederick, MD
Contact
For more information, email henry_levin@nih.gov or visit sete.nichd.nih.gov.