



You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Gabriella Finkielstain, MD, Postdoctoral Fellow
- Patricia Forcinito, BS, Special Volunteer
- Julian Lui, PhD, Postdoctoral Fellow
We investigate the cellular and molecular mechanisms governing childhood growth and development. We are especially interested in the mechanisms that allow rapid proliferation and hence rapid body growth in young mammals and that subsequently suppress proliferation, thus setting a fundamental limit on the adult body size of the species. One goal of this work is to improve our understanding of childhood growth disorders. In addition, by elucidating the physiological mechanisms that govern normal cell proliferation, we seek to understand better the pathological mechanisms that allow abnormal cell proliferation particularly in childhood cancers.
Cellular mechanisms limiting childhood growth
Organ formation and development during mammalian embryogenesis are orchestrated by complex changes in gene expression. These embryonic genetic programs have been studied intensively. However, mammalian development does not cease at birth; maturation continues during postnatal life in multiple tissues. The genetic mechanisms required for such postnatal development have received far less attention.
In particular, little is known about the mechanisms that slow somatic growth. Mammalian body growth is rapid in early postnatal life, but then the growth rate slows, eventually declining toward zero, as the organism approaches its adult body size. This decrease in growth rate is due primarily to a decline in the rate of cell proliferation. We recently showed that this decline in proliferation does not reflect an increase in average cell-cycle time but rather a decrease in the growth fraction, that is, the number of cells that are actively proliferating (1). One clue to the underlying mechanism is that the decline in proliferation occurs simultaneously in multiple tissues but does not appear to be orchestrated by a hormonal or other systemic mechanism.
We therefore hypothesized that the coordinated growth deceleration is directed by a program of gene expression that is common to multiple tissues. To test this hypothesis, we used microarray analysis to identify changes in gene expression that occur during early postnatal life (2). We found that extensive changes in gene expression occur with age in mice during the period of postnatal growth. In each of the organs studied—kidney, lung, and heart—more than 2000 genes were found to be significantly upregulated with age and more than 2800 genes downregulated. The number of genes that were regulated coordinately in all three organs was strikingly high, with 1207 genes downregulated in all three organs and 428 upregulated in all three organs—far more overlap than would be expected by chance. These findings suggest that there is a complex program of gene expression common to multiple organs occurring during postnatal growth, in addition to the expected tissue-specific changes. The common program included genes involved in regulating G1/S and G2/M checkpoints, Hedgehog signaling, and Wnt/ß-Catenin signaling (2). Many of the most strongly regulated genes were imprinted (3). In these pathways, more genes were downregulated with age than were upregulated.
We then focused on genes that were up- or downregulated in all three organs and thus more likely to contribute to the putative common program of growth deceleration (2). For more detailed characterization, we selected five genes, Igf2, Mest, Peg3, Sox4, and Igf2bp3, that were markedly downregulated in all three organs. To better quantify the temporal changes in gene expression identified by microarray, we measured mRNA expression levels by real-time PCR in the same three organs studied by microarray. The real-time PCR results confirmed the marked decline in mRNA expression with age. We also measured mRNA levels in a fourth organ, liver, and found dramatic reductions with age, with a pattern very similar to those observed in heart, kidney, and lung, further confirming that this gene expression program is occurring globally in multiple organs. Thus, the common program of gene expression was observed in organs of widely differing embryonic origin, specifically, kidney and heart, which are derived from mesoderm, and lung and liver, which are derived from endoderm.
We then determined which cell types in each organ expressed Igf2, Mest, and Peg3 in the 1-week-old mouse by in situ hybridization (2). In general, we found expression in organ-specific parenchymal cells, suggesting that the coordinate decline in expression in multiple organs is not occurring simply because gene expression is restricted to cells that are common to multiple organs, such as endothelial cells or stromal fibroblasts. Instead, expression of these genes appears to be declining coordinately in multiple cell types throughout the body, suggesting a common genetic program in many tissues coordinately.
We next performed in situ hybridization in tissues from 4-week-old mice. We had considered the possibility that some cells might continue to show high levels of expression at four weeks of age but that there might be fewer of these expressing cells than at one week of age, thus explaining the overall declines in expression observed by microarray and real-time PCR. Such a finding would support the hypothesis that, with age, many cells stop expressing these growth-promoting genes and thus perhaps drop out of the cell cycle, while a dwindling number continue to express these genes and continue to proliferate. However, our findings did not support that hypothesis. Instead, we found that, at four weeks of age, mRNA expression was uniformly low for all genes studied in all organs, indicating that the overall decrease in Igf2, Mest, and Peg3 expression was due to lower expression per cell rather than fewer expressing cells.
The observed reductions in expression of Igf2, Mest, and Peg3 are likely to contribute to postnatal growth deceleration. Igf2 encodes an insulin-like growth factor. IGF-II acts primarily in a paracrine fashion, binding to the type1 IGF receptor. The liganded receptor acts as a tyrosine kinase to potently stimulate proliferation in many cell types, both in vivo and in vitro. Targeted ablation of Igf2 in mice leads to marked growth retardation, with body mass 60% of normal at birth. Postnatal growth maintains the body mass at 60% of normal. Thus ablation primarily affects somatic growth in early life, which fits the finding that Igf2 expression occurs only during early life and declines thereafter. Conversely, overexpression of Igf2 in vivo leads to increased somatic growth. Mest encodes a presumptive a/b fold hydrolase, with unknown substrates. Targeted ablation causes embryonic growth retardation. Conversely, in mouse interspecies hybrids, loss of imprinting of Mest is associated with increased fetal somatic growth. Peg3 encodes a nuclear protein that contains 12 Kruppel-type zinc finger domains and two proline-rich repeat domains and is thought to participate in apoptotic pathways. Targeted ablation of Peg3 produces mice with body mass 85% of wild type at E17.5, 81% at birth, and 65% by four weeks of age. The major organs in the mutants, although proportionally smaller, are morphologically normal. Thus, Igf2, Mest, and Peg3 are each required for normal embryonic growth, suggesting that the decline in expression during postnatal life contributes to postnatal growth deceleration.
We also asked whether the observed changes in gene expression are a function of time per se or a function of growth. In rats, PTU-induced hypothyroidism and growth retardation during the first 5 weeks of life delayed the declines in expression of Igf2, Mest, and Peg3, the three genes implicated in growth regulation. These data are consistent with the hypothesis that the normal decline in expression of these genes is driven, not by age per se, but rather by the process of growth; therefore a prior period of growth inhibition slows this decline. We previously showed that changes in gene expression in the growth plate are driven by growth rather than time per se (4). Therefore, our findings indicate that a genetic program driven by body growth represents a general mechanism for regulating somatic size.
Together, these findings suggest the following model to explain the deceleration and eventual cessation of somatic growth in mammals. In late embryonic and early postnatal life, a network of growth-promoting genes is expressed at high levels. The resulting growth causes the expression of these growth-promoting genes to decline, which in turn slows the rate of growth. Eventually, their expression levels decline sufficiently to cause somatic growth to cease. If some external condition, such as hypothyroidism transiently restricts growth, then the slow growth slows the decline in gene expression, thus preserving future growth capacity. Following the period of growth inhibition, the expression levels of the growth-promoting genes will be higher than normal, and consequently the growth rate will be greater than normal. Thus the model provides an explanation for the phenomenon of catch-up growth, which is defined as a growth rate that is greater than normal for age following a period of growth inhibition.
In summary, our recent findings point to an extensive genetic program during the postnatal period involving upregulation and downregulation of thousands of genes. Some appear to be organ-specific, but many are regulated in a concerted fashion in multiple organs. Some of these common genes regulate cell proliferation and thus may constitute part of the mechanism that causes somatic growth to slow and eventually cease. We found evidence that several of the genes likely to participate in this growth-limiting program—Igf2, Mest, and Peg3—are themselves regulated by growth, suggesting that, in the embryo, a gene expression pattern is established that allows for rapid somatic growth of multiple tissues; but then, during postnatal life, this growth leads to negative-feedback changes in gene expression that in turn slow and eventually halt somatic growth, thus imposing a fundamental limit on adult body size.
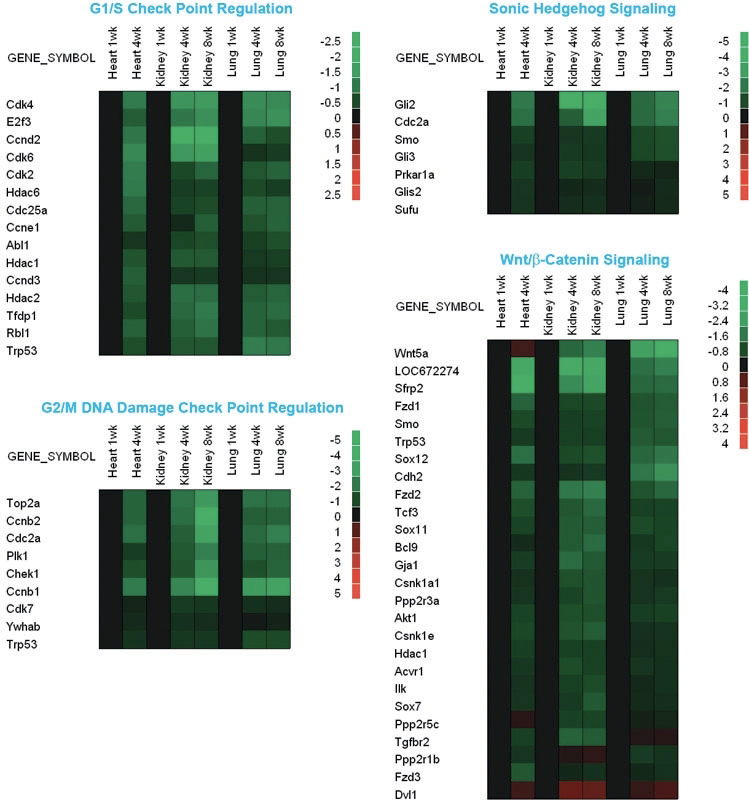
Figure 1. Changes in gene expression in mouse lung, kidney, and heart during early postnatal life
Microarray analysis was used to evaluate changes in gene expression in lung and kidney of 1-, 4-, and 8-week-old mice and in heart of 1- and 4-week-old mice. Green rectangles represent down-regulated genes; red rectangles represent up-regulated genes compared with 1-week-old animals; black rectangles represent no change in gene expression.
Publications
- Chang M, Parker EA, Muller T, Haenen C, Mistry M, Finkielstain G, Murphy-Ryan M, Barnes KM, Sundaram R, Baron J. Changes in cell cycle kinetics responsible for limiting somatic growth in mice. Pediatr Res 2008 64:240-245.
- Finkielstain GP, Forcinito P, Lui JC, Barnes KM, Marino R, Makaroun S, Nguyen V, Lazarus JE, Nilsson O, Baron J. An extensive genetic program occurring during postnatal growth in multiple tissues. Endocrinology 2009 150:1791-1800.
- Lui JC, Finkielstain GP, Barnes KM, Baron J. An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am J Physiol: Regul Integr Comp Physiol 2008 295:R189-196.
- Marino R, Hegde A, Barnes KM, Schrier L, Emons JA, Nilsson O, Baron J. Catch-up growth after hypothyroidism is caused by delayed growth plate senescence. Endocrinology 2008 149:1820-1828.
Collaborator
- Ola Nilsson, MD, PhD, Karolinska University Hospital, Stockholm, Sweden
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.