



You are here: Home > Section on Molecular Endocrinology
Receptors and Actions of Peptide Hormones and Regulatory Proteins in Endocrine Mechanisms

- Maria L. Dufau, MD, PhD, Head, Section on Molecular Endocrinology
- Masato Fukushima, MD, PhD, Adjunct Scientist
- Junghoon Kang, PhD, Postdoctoral Fellow
- Mingjuan Liao, PhD, Staff Fellow
- Xiolan Sun, PhD, Postdoctoral Fellow
- Chon-Hwa Tsai-Morris, PhD, Staff Scientist
- Joaquin Villar, PhD, Postdoctoral Fellow
We investigate the molecular basis of peptide hormone control of gonadal function, with particular emphasis on the structure and regulation of the luteinizing hormone receptor (LHR) and prolactin (PRL) receptor (PRLR) genes. We also investigate the regulatory mechanism(s) involved in the progress of spermatogenesis and the control of Leydig cell function. Our studies focus on the regulation of human LHR transcription (nuclear orphan receptors, epigenetics, DNA methylation, and second messengers) as well as on the multiple-promoter control of hPRLR gene transcription. We are elucidating the function of two inhibitory short forms of the prolactin receptors and their relevance to physiological regulation and breast cancer. We also investigate novel gonadotropin-regulated genes of relevance to the progression of testicular gametogenesis, Leydig cell function, and other endocrine processes.
The luteinizing hormone receptor (LHR)
Characterization of regulatory mechanisms revealed that human LHR (hLHR) expression is subject to regulation by a complex network and the participation of various signaling cascades through the gene's promoter region at both genetic and epigenetic levels. The histone deacetylase HDAC1/2/mSin3A complex was shown to be recruited to the proximal Sp1 binding site to elicit chromatin-localized condensation and silencing of the LHR gene. The status of DNA methylation and histone acetylation in the LHR promoter region operates coordinately to orchestrate the transcriptional LHR level. The PI3K/PKCzeta signaling pathway was shown to be critical for trichostatin A (TSA)–induced LHR transcriptional activation through phosphorylation of Sp1 at Ser641 and release of p107, which interacts with Sp1. LHR transcriptional activation is initiated by changes in chromatin structure induced by TSA that causes the release of phosphatase PP2A in JAR cells or of PP1 in MCF-7 cells; the phosphatase is associated with Sp1 directly or through HDAC 1/2, respectively, at the promoter (Zhang et al.). Other recent studies also demonstrated a critical role of the PKCalpha/Erk cascade in PMA-induced activation of LHR gene through Sp1 phosphorylation (other than Ser641 site), which causes release of the HDAC1/mSin3A repressor complex anchored at Sp1 sites (Liao et al.). However, HDAC2 from the complex did not dissociate from the promoter, indicating distinct roles of HDAC1 and HDAC2. The phosphorylation induced by PKCalpha/ERK signaling disrupts the interaction between Sp1 and HDAC1, which causes release of HDAC1/mSin3A complex to activate hLHR. This is consistent with our finding that mSin3A functions as corepressor for HDAC1 but not HDAC2 in the regulation of LHR gene expression. In other studies we investigated the participation of PC4, a potential coactivator of Sp1, in the transcriptional control of LHR expression. Knockdown of PC4 expression with its specific siRNA significantly reduced LHR promoter activity in MCF-7 and JAR cancer cells. PC4 synergistically enhanced Sp1-activated gene expression, indicating a direct or indirect interaction of PC4 and Sp1. These studies have demonstrated that PC4 acts as a coactivator of Sp1 to modulate LHR transcription.
Gonadotropin-regulated genes
Gonadotropin-regulated long chain fatty acid acyl CoA synthase (GR-LACS), identified in our laboratory, is a member of the LACS family regulated by LH/hCG in rat Leydig cell (LC). It has been suggested by others that the mouse/human homologs lipidosin/bubblegum participate in X-linked adrenoleukodystrophy (X-ALD), a disorder with accumulation of very long chain fatty acids (VLCFA) in tissues and blood. GRLACS/lipidosin null mice generated in our laboratory did not support GR-LACS's association with X-ALD. The studies revealed a role of GR-LACS in reducing the aging process of the LC and its participation in gonadotropin-induced testicular desensitization of testosterone production that is observed in the adult animals (Sheng et al.).
Gonadotropin-regulated testicular helicase (GRTH/Ddx25), a multifunctional enzyme discovered in our laboratory, is an essential regulator of sperm maturation. GRTH null mice are sterile due to spermatid arrest and failure to elongate. A striking apoptosis was observed in spermatocytes entering the metaphase of meiosis in the null mice. This indicated the important role of GRTH in determining the survival and apoptotic fate of adult germ cells. Despite apoptosis, the progress of the remaining viable meiotic cells (about 30%) to the point of arrest in haploid cells (step 7-8 of spermatids) was unimpaired, presumably due to the activation of survival mechanisms and/or compensatory participation of other helicases. We found that GRTH prevents germ cell apoptosis by regulating pro- and anti-apoptotic factors/pathways in germ cells (mitochondrial, death receptor, NF-kappaB) (Gutti et al.). Through its association with CRM-1, GRTH participates in the export of selected messages relevant to the progress of spermatogenesis from the nuclear to cytoplamic sites, including the Chromatoid Body (CB) of spermatids (equivalent to somatic P bodies; contains members of the RNA-induced silencing complex [RISC] and is viewed as a storage/processing site of mRNAs during spermatogenesis) and polyribosomes, where GRTH participates in translation of proteins that are essential for spermatogenesis. In recent studies, we investigated the nuclear/cytoplasmic shuttling of GRTH in germ cells and its impact on the structure of the CB of spermatocytes in cell culture, using immunofluorescence and electron microscopy. GRTH resides in the nucleus, the cytoplasm, and the CB. Treatment of these cells with inhibitors of nuclear export caused nuclear retention of GRTH and its absence from the cytoplasm and CB. On other hand, proteins of the RISC complex that do not participate in mRNA transport and reside in the CB and cytoplasm were excluded from the CB and accumulated in the cytoplasm upon treatment with the inhibitor. This also occurred in spermatids of GRTH KO mice. The structure of the CB changes from lobular-filamentous to a small condensed entity after inhibitor treatment, resembling the CB of the KO. GRTH did not interact with RISC members. Owing to its export/transport function, GRTH is essential for the transport of messages to the CB, which governs the CB structure in spermatids, and for maintaining systems that participate in mRNA storage and their processing during spermatogenesis.
Prolactin receptor (PRLR)
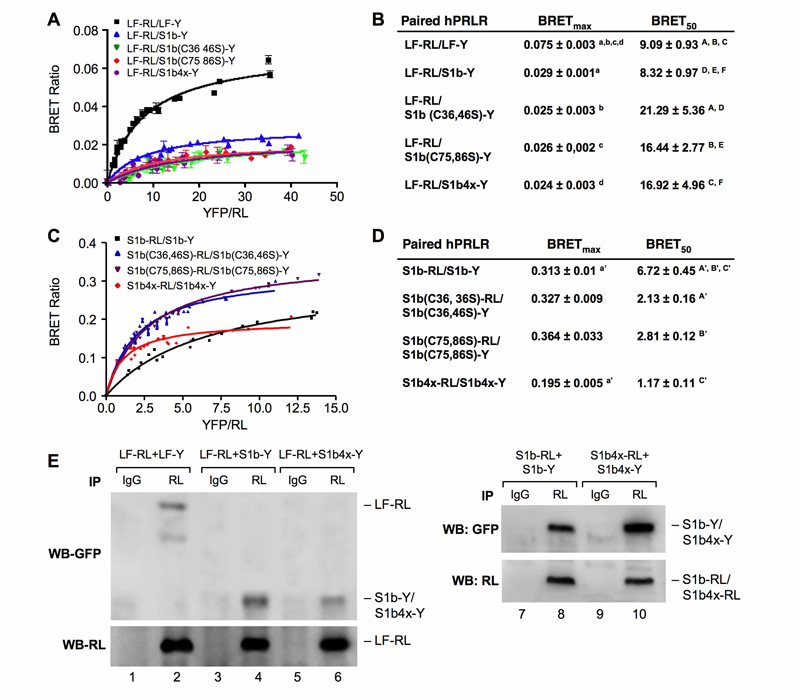
Click for a larger view.
Figure 1. A-D: BRET saturation curve of hetero-and homo-dimerization of wild-type and mutated hPRLR variants. E: Effect of S1b Cys mutants on the formation of homodimer and heterodimerizatin with LF.
A-D: HEK293 cells were cotransfected with a constant (A) LF-RL (0.2µg) or (C) wild-type or mutated S1b-RL (0.2 µg) with increasing DNA concentration of YFP fusion construct (LF-Y, wild-type S1b-Y or mutant S1b-Y), respectively. In all cases the amount of the YFP constructs (determined by fluorescence levels) was similar at each designated dose. RL: Renilla luciferase; Y, Aequora enhanced YFP. The BRET ratio, total luminescence, and total fluorescence were measured after transfection. BRET levels were plotted as a function of the ratio of the expression level of YFP construct (quantitated by total fluorescence of cells) over RL construct (quantitated by luminescence of cells [YFP/RL]). This ratio reflected the fold-change at the corresponding receptor expression level. The results are representative of three independent experiments carried out in triplicates. S1b(C#, #S): mutation of cysteine pair to serine occurred at the designated amino acid location (#). S1b4x:Cys to serine mutation at amino acids #36, 46, 75 and 86 of the short form S1b. (B) and (D): Parameters derived from BRET saturation curve of hetero- and homo-dimerization of wild-type and mutated hPRLR variants (A & C). BRETmax is the maximal BRET ratio obtained for a given pair. The BRET50 represents the relative affinity as acceptor/donor ratio required to reach half-maximal BRET values. Identical superscripts indicate a statistical significance between experimental groups, p < 0.01. E: CO-IP analysis of transfected receptor wild-type (LF-RL, LF-Y, S1b-RL, S1b-Y) and mutant (S1b4x_RL, S1b4X-Y) into HEK293 cells. Immunoprecipitation (IP) with RL antibody. IgG (negative control). WB (Western blot) with GFP or RL antibody. WB with RL antibody was used as loading control (IP) (lower panels).
Structure/function: Our early studies demonstrated: (1) the presence of long (LF) and short forms (SF) of PRL; (2) formation of homo- and heterodimers of these forms in the absence of hormone and the action of PRL as a conformational modifier that induces activation of the JAK2/STAT5 signaling through LF and of JAK2 signalling through SF; (3) the short form of the PRLR (S1b) with an abbreviated cytoplasmic tail silences PRL-induced activation of gene transcription by the long-form. Mutation of any one of the two paired cysteines in S1b (S1bx), residing in extracellular subdomain 1 (D1), eliminated the inhibitory action of S1b. The constitutive JAK2 phosphorylation observed in S1b was not present in cells expressing S1bx due to disruption of JAK2 association (intracellular conserved proline-rich domain at the Box1 docking site for JAK2). BRET50 showed lower LF/S1bx than LH/S1b heterodimeric association and increased affinity in S1bx homodimerization (Figure 1), favoring homo-dimerization of LF and PRLR-induced signaling. Computer modeling based on PRLR crystal structure (extracellular) showed that minor changes in the tertiary structure of D1 upon S-S disruption propagated to the quaternary structure of the homodimer, affecting the dimerization interface. Wild-type dimers were stabilized only through interactions between H-bonds in D2 subdomains, while the mutant developed additional H-bonds that bridged D1s (Figure 2). These changes explain the higher homodimerization affinity of the mutant (S1bx) and provide the structural basis for its lack of inhibitory function (Xie et al.). The PRLR conformation, as stabilized by S-S bonds, is required for the inhibitory action of S1b on PRL-induced LF-mediated function and JAK2 association/activity.
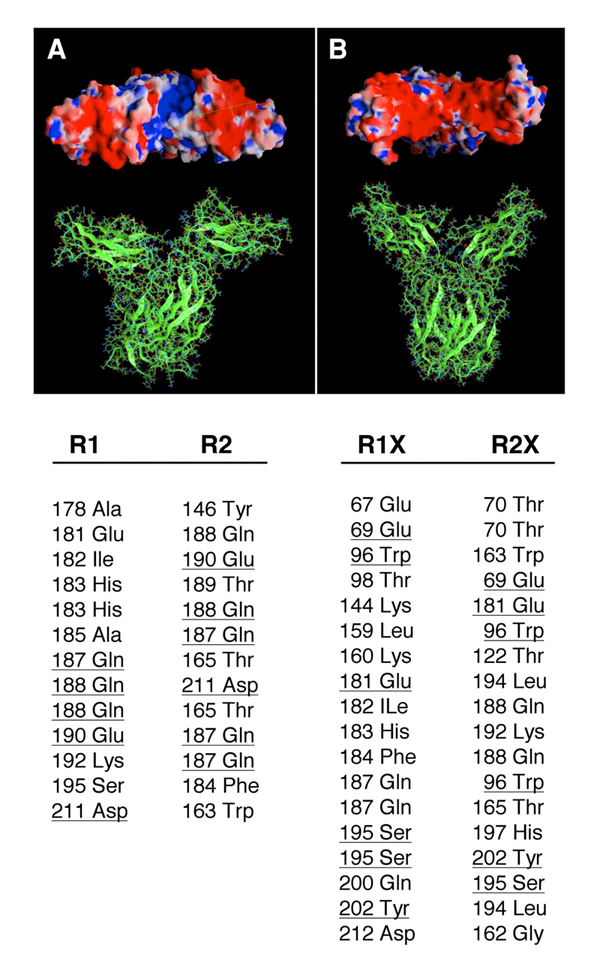
Click for a larger view.
Figure 2. Molecular models of the wildtype (A) and double mutant (B) EC domains of the hPRLR dimers.
In the wild type, the intermolecular hydrogen bonds (HB) network involves only the D2 subdomains, while both D1 and D2 subdomains are H-bonded in the C36S, C46S, C75S, and C86S mutant. Electrostatic potentials on the molecular surfaces of the PRLR dimers are shown in the upper panels (viewed from the EC side towards the membrane plane). In the wild type, a groove of positive potential (blue) is flanked by two negative regions (red); this positive crevice is occluded in the mutant receptor by the D1 subdomains that are now H-bonded. The lower panel lists the corresponding inter-monomer HB of the EC domains, as obtained in the molecular dynamics simulations of the receptors. The wild-type dimer contains 13 HB pairs, involving 16 aa, with 4 aa common to both monomers (underlined); the mutant dimer contains 18 HB pairs, involving 25 aa, with 5 aa common to both monomers (underlined). R1 and R2 denote the wild-type monomers; R1X and R2X denote the mutant monomers. Many of these inter-monomer HBs involve at least one backbone atom.
Transcriptional studies. Transcription of the human PRLR is governed by an estradiol (E2)/estrogen receptor (ER)-alpha mechanism independent of an estrogen-responsive element. Complex formation of E2/ER-alpha with SP1 and C/EBPbeta that associate to cognate elements leads to coactivator assembly and recruitment of TFIIB and Pol II. We are characterizing the physical interaction domains involved in the complex association among transfactors. This study provides evidence for direct association of components of the complex essential for PRLR transcription through defined interaction domains for the individual proteins.
Publications
- Gutti R, Tsai-Morris C-H, Dufau ML. Gonadotropin regulated testicular helicase (DDX25) an essential regulator of spermatogenesis, prevents testicular germ cell apoptosis. J Biol Chem 2008 283:17055-17064.
- Liao M, Zhang Y, Dufau ML. Protein kinase C alpha-induced derepression of the human luteinizing hormone receptor gene transcription through ERK-mediated release of HDAC1/Sin3A repressor complex from Sp1 sites. Mol Cell Endocrinol 2008 22:1449-1463.
- Sheng Y, Tsai Morris C-H, Li J, Dufau ML. Lessons from the gonadotropin-regulated long chain acyl-CoA synthetase (GRLACS) null mouse model: a role in steroidogenesis, but not result in X-ALD phenotype. J Steroid Biochem Mol Biol 2009 114:44-56.
- Xie YL, Hassan SA, Qazi AM, Tsai-Morris CH, Dufau ML. Intramolecular disulfide bonds of the prolactin receptor short form are required for its inhibitory action on the function of the long form of the receptor. Mol Cell Biol 2009 29:2546-2555.
- Zhang Y, Liao M, Dufau ML. Unlocking repression of the human LH receptor gene by trichostatin A-induced cell-specific phosphatase release. J Biol Chem 2008 283:24039-24046.
Collaborator
- Sergio A. Hassan, PhD, Center for Molecular Modeling, CIT, NIH, Bethesda, MD
Contact
For more information, email dufau@helix.nih.gov.