


You are here: Home > Section on Epigenetics and Development
The X-chromosome and Gender Effects in Physiology, Pathophysiology, and Longevity

- Carolyn Bondy, MD, Head, Section on Epigenetics and Development
- Vladimir Bakalov, MD, Staff Clinician
- Clara Cheng, PhD, Biologist
- Jian Zhou, MD, PhD, Staff Scientist
- Harley Gould, BS, Postbaccalaureate Fellow
- Alexis Merz, BS, Postbaccalaureate Fellow
Many obvious phenotypic differences between normal women and men result from differential exposure to sex steroids. However, exposure to sex steroids does not adequately explain important differences in gender-specific susceptibility to disease and longevity. We propose that differential X-chromosome gene dosage contributes to fundamental biological differences between females and males. While researchers had considered the second X-chromosome in female cells totally inert as a result of random X-inactivation, the distinct phenotype in 45,X females with Turner syndrome (TS) indicates that the second X-chromosome is important for normal female development. Epigenetic mechanisms are involved in regional X-inactivation as well as in genomic imprinting and thus contribute to sex differences by X-chromosome gene-dosage effects. Our research aims to identify and define the function of X-chromosome genes involved in the differential development and function of brain, reproductive, metabolic, and immune systems in women and men. Studying girls and women with TS provides a unique opportunity to elucidate X-chromosome gene-dosage effects and deepen our understanding of TS, which affects approximately 1 in 2,000 females. Our research will improve our ability to care for girls and women with TS and will enhance our understanding of women's increased susceptibility to autoimmune disease and men's elevated risk for coronary disease.
X-chromosome, genomic imprinting, and longevity
Genomic imprinting involves the selective expression, determined by parental origin, of certain genes, often associated with DNA methylation of imprinted, or silenced, alleles. Genomic imprinting of X-linked genes causes different gene expression in females and males, given that normal women are mosaic for maternally and paternally inherited active X-chromosomes (XM and XP) while men are monosomic for XM. Imprinted XM genes would still be expressed in about 50 percent of female cells but not in male cells.
Women enjoy greater longevity than men mainly because of their lower risk, across all age groups, for ischemic heart disease. Women's primary advantage in this regard is their salutary "gynoid" fat distribution, meaning adipose tissue preferentially concentrated subcutaneously in the hips and thighs. In contrast, normal men tend to concentrate fat in the abdominal area, especially intraperitoneal visceral fat, which has many adverse metabolic effects, including an atherogenic lipid profile and elevated mediators of inflammation, which cause coronary and cerebral artery atherosclerosis. To test the hypothesis that X-chromosome genomic imprinting contributes to the regional fat and metabolic differences between normal women and men, we studied the risk factors in groups of women monosomic for XM versus XP. The XM and XP groups had a similar BMI and total body fat, but women with XM had greater abdominal and intra-abdominal visceral fat, which were associated with higher LDL-cholesterol and triglycerides than in the XP group. Our finding of a male-type fat distribution and lipid profile in XM women supports the view that differential X-chromosome gene dosage, determined by genomic imprinting, contributes to the excess mortality from ischemic heart disease in 46,XMY men.
Given the different parental origins of the X-chromosome in males and females, genomic imprinting is predictably associated with differential effects depending on the sex of the offspring (in contrast to autosomal imprinting). Thus, X-linked imprinting is not expected to regulate traits that are not sexually dimorphic, such as renal or cardiac development. We proved our prediction in a recent study showing equal prevalence of renal and cardiovascular defects in XM versus XP groups with TS. In contrast, somatic size is sexually dimorphic, with mentypicallyconsiderably larger than women; in fact, we have shown that size is related to X origin.
Congenital cardiovascular defects in Turner syndrome
Congenital cardiovascular disease may be the most serious medical problem in monosomy X or TS. Pioneering the use of high-resolution magnetic resonance angiography (MRA) for TS, we demonstrated cardiovascular anomalies in about 50% of study subjects, in contrast to the previously accepted 20–30% prevalence based on echocardiographic studies. Whereas congenital heart defects in TS were thought to be limited to left-sided, outflow tract defects, MRA revealed a high prevalence of major venous malformations, including partial anomalous pulmonary venous return and persistent left superior vena cava affecting over 20% of the study population. The most common anomaly disclosed by our study was a distinctive aortic deformation affecting about 50% of women with TS, termed elongated transverse arch of the aorta (ETA). This newly described aortic abnormality seems to be predictive of aortic complications and hence mandates vigilant surveillance of aorta dimensions in affected yet asymptomatic patients.
In recent years, we have seen frequent case reports of fatal aortic dissections in relatively young women with TS. The incidence was unclear, and it was unknown whether dilatation of the ascending aorta predicted aortic dissection in TS and, if so, what specific aortic diameters should provoke concern. In other patient groups at risk for dissection such as Marfan syndrome, dilatation of the ascending aorta is a predictor of an acute aortic event. Thresholds of 5.5–6 cm for the general population and 5 cm for those with Marfan syndrome guide "prophylactic" intervention to replace or stabilize the aneurysmal segment. Under this "one size fits all" prescription, however, women (who are smaller than men, for whom the guidelines were developed) have a higher likelihood of dissection/rupture and death. Accordingly, it has been suggested that intervention at a smaller diameter would save more female lives. Given that aortic diameter is closely correlated with somatic size, the aorta should be smaller than "normal" in very petite women with TS; such women might actually have significant aortic dilatation at diameters considered in the normal range.
We recently reported the first prospective measure of the incidence of aortic dissection in TS and proposed new guidelines to identify high-risk patients (Matura et al., Circulation 2007;116:1663). We evaluated aortic diameters and other parameters in a large group of asymptomatic, unselected women with TS and followed the women for an average of three years. During the period, we recorded three cases of aortic dissection among 158 patients, translating into an incidence of about 618 cases per 100,000 TS years compared with about 1 per 100,000 non–TS women years. The women who dissected were in their 40s and had aortic diameters from 3.7–4.8 cm. They were under cardiologist care in their home area, but, because their aortic diameter was not more than 5 cm, they were not considered candidates for prophylactic intervention. Hence, it is inadvisable to wait for aortic diameter to reach 5 cm in TS patients. We investigated various methods of normalizing MRI-measured aortic diameters to individual body size and found that aortic diameter/body surface area (Aortic Size Index, ASI) provided the highest correlation and greatest accuracy in identifying those at risk for dissection. In summary, 25% of women with absolute ascending aortic diameter greater 3.5 cm and 33% of women with ASI greater than 2.5 cm/m2 experienced aortic dissection within approximately three years of follow-up.
Thus, for screening purposes, we suggest use of the ASI 95th percentile of 2 cm/m2 for the ascending aorta determined by MRI, CT, or cardiac echo. This measure takes into account the considerable size variation of patients and identifies about 30 percent of women with TS who require close monitoring. If the ASI is equal to or greater than 2.5 cm/m2, the patient should be evaluated for prophylactic intervention. Clearly, further study is needed to determine whether a beta blocker or renin-angiotensin system blockade may prevent or retard aortic dilatation in TS patients and whether prophylactic surgery may reduce the incidence of aortic dissection and rupture.
This year, we received a Bench-to-Bedside Award for a randomized treatment study comparing angiotensin converting enzyme inhibitor vs. angiotensin 1 receptor blocker, based on preliminary data shown in the figure below:
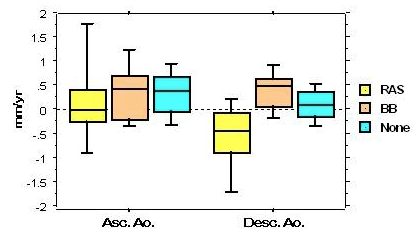
Effects of beta blocker vs. renin-angiotensin blocker on aortic diameter changes in Turner syndrome
BB, beta-blocker; RAS, renin-angiotensin blocker; Asc.Ao., ascending aorta; Desc.Ao., descending aorta
The X-chromosome, ovarian failure, and psychosocial function
Previous small surveys and anecdotal reports indicated that many girls and women with TS suffer from shyness and social anxiety. This "shyness trait" was traditionally attributed to stigmatization because of short stature (Cardoso et al., Gynecol Endocrinol 2004;19:313). More recently, researchers suggested that social difficulties could be neurobiologically based, caused by genomic imprinting of X-linked genes involved in "social cognition." We showed that women with TS experience a rate of life-time depression similar to women from gynecologic clinic samples, with no "autistic spectrum" diagnoses. In open-ended interviews, girls and women identified ovarian failure, infertility, or "gender identity" as major issues related to their experience of TS. Thus, our findings suggest that the experience of ovarian failure is a major hurdle for older girls and women with TS while major psychological diagnoses and autistic-type disorders are uncommon.
To investigate the behavioral impact of early ovarian failure, we compared psychosocial functioning in women with TS and women with 46,XX premature ovarian failure. We reasoned that, if the experience of premature ovarian failure per se leads to specific difficulties with social interactions in young women, then the two groups should demonstrate similar responses on tests of psychosocial function. Women with normal ovarian function served as contemporaneous controls. Our two ovarian failure groups reported similar increases in shyness and social anxiety and lower self-esteem than did women with normal ovarian function. The findings suggest that the shyness and poor self-esteem observed in women with TS reflect the experience of premature ovarian failure. The feelings of social inadequacy are associated with social isolation, difficulty in partnering, and very limited sexual functioning. Current studies target the role of infertility, childlessness, sex steroid effects, or altered body image in impaired social functioning in women with TS. We will also address the issue of how best to inform a young woman and her family about the diagnosis of premature ovarian failure so as to minimize the psychological trauma. In addition, we plan to extend our studies to girls and young women who have survived cancer treatments that compromise ovarian function.
Publications
- Sachdev V, Matura LA, Sidenko S, Ho VB, Arai AE, Rosing DR, Bondy CA. Aortic valve disease in Turner syndrome. J Am Coll Cardiol. 2008;51:1904-1909.
- Bakalov VK, Cheng C, Zhou J, Bondy CA. X-chromosome gene dosage and the risk of diabetes in Turner syndrome. J Clin Endocrinol Metab. 2009;94:3289-96.
- Wooten N, Bakalov VK, Hill S, Bondy CA. Reduced abdominal adiposity and improved insulin sensitivity in GH-treated girls with Turner syndrome. J Clin Endocrinol Metab. 2008;93:2109-14.
- Bondy CA, Cheng C. Monosomy for the X Chromosome. Chromosome Res. 2009;17:649-58.
- Bondy CA. Aortic Dissection in Turner Syndrome. Current Opinion in Cardiology 2009;23:519-26.
Collaborators
- Andrew Arai, MD, Laboratory of Cardiac Energetics, NHLBI, Bethesda, MD
- Jeff Baron, MD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Barbara Biesecker, MS, Social and Behavioral Research Branch, NIHGR, Bethesda, MD
- Graça Cardoso, MD, PhD, Behavioral Endocrinology Branch, NIMH, Bethesda, MD
- Andrew Griffith, MD, PhD, Neuro-Otology Branch, NIDCR, Bethesda, MD
- Suvimol Hill, MD, Department of Radiology, Clinical Center, NIH, Bethesda, MD
- Vince Ho, MD, Department of Radiology, Clinical Center, NIH, Bethesda, MD
- James Reynolds, MD, Nuclear Medicine, Clinical Center, NIH, Bethesda, MD
- Douglas Rosing, MD, Cardiology Branch, NHLBI, Bethesda, MD
- Judith L. Ross, MD, Thomas Jefferson University, Philadelphia, PA
- Vandana Sachdev, MD, Cardiology Branch, NHLBI, Bethesda, MD
- Peter Schmidt, MD, Behavioral Endocrinology Branch, NIMH, Bethesda, MD
- Constantine Stratakis, MD, D(med)Sci, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Erica Sutton, MS, Social and Behavioral Research Branch, NIHGR, Bethesda, MD
- James Troendle, PhD, Biometry and Statistics Branch, NICHD, Bethesda, MD
- Jack A. Yanovski, MD, PhD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
Contact
For further information, contact bondyc@mail.nih.gov.