


You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Patricia Forcinito, BS, Special Volunteer
- Julian Lui, PhD, Postdoctoral Fellow
- Sao Fong Cheung, Predoctoral Fellow
- Angela Delaney, MD, Clinical Fellow
- Fariha Kamran, MD, Clinical Fellow
We investigate the cellular and molecular mechanisms governing childhood growth and development. We are especially interested in mechanisms that allow rapid cell proliferation and hence rapid body growth in young mammals and subsequently suppress proliferation, thus setting a fundamental limit on the adult body size of the species. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure and overgrowth. In addition, further investigation of the identified growth-limiting mechanisms may lead to broader medical applications, because disruption of these mechanisms may contribute to oncogenesis, and conversely transient therapeutic suspension of growth-limiting mechanisms in adult cells might be used to achieve tissue regeneration.
Cellular mechanisms limiting childhood growth
The human fetus grows at an enormous rate, increasing in mass more than 100 fold between the end of embryogenesis and birth. Somatic growth then slows progressively in postnatal life and ceases by the end of the second decade. The deceleration in body growth is due primarily to a progressive decline in cell proliferation, but the underlying mechanisms remain largely unknown.
One clue regarding the mechanism is that growth deceleration occurs coordinately in multiple organs in order to maintain body proportions; yet this coordination does not appear to be orchestrated by a systemic mechanism. We therefore hypothesized that postnatal growth deceleration results from a genetic program that occurs simultaneously in multiple tissues. Consistent with this hypothesis, we identified an extensive program of gene expression that occurs between 1 and 8 weeks of age simultaneously in lung, kidney, and heart of mice (see reference 1). The common program included genes involved in regulating G1/S and G2/M checkpoints, Hedgehog signaling, and Wnt/β-Catenin signaling (see reference 1). We found that many of the most strongly regulated genes are imprinted. We subsequently showed that this multi-organ, postnatal genetic program has remained highly conserved in the approximately 20 million years since mice and rats diverged (Figure 1A, 1B), providing strong confirmation of the validity and biological importance of the program (see reference 2).
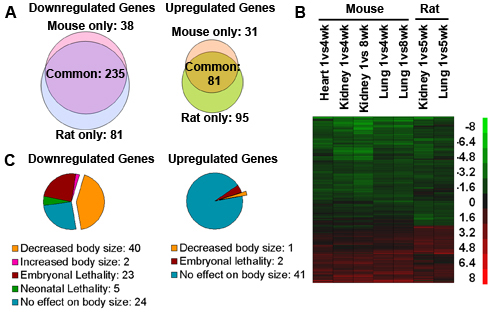
Figure 1. A multi-organ postnatal genetic program common to mice and rats.
A) Venn diagrams from microarray analysis showing the number of genes significantly downregulated and upregulated with age in mouse versus rat. Analysis included 532 genes that showed age regulation (P < 0.05; ≥2.0-fold) in all mouse organs studied (kidney, lung, heart) or both rat organs studied (kidney, lung). B) Heat maps based on microarray analysis of the same 532 genes. Each row corresponds to a gene, ranked by hierarchal clustering. Green: downregulation with age; red: upregulation. Scale values are log2(fold difference). C) Pie chart depicting knockout mouse phenotypes of 316 genes that showed uniform age regulation (≥2.0-fold, P< 0.05) in all organs of both species.
Multiple lines of evidence suggest that at least part of this program contributes to body growth deceleration. For the downregulated genes in the program, gene ontology analyses indicated strong overrepresentation of genes implicated in cell growth/ proliferation (see reference 2). Furthermore, of the knockout phenotypes that have been reported for age-downregulated genes, more than one third showed a decrease in body size without any detected underlying disease, implying that these age-downregulated genes in the program promote somatic growth (Figure 1C). This concept was further supported by in vitro studies using siRNA-mediated knockdown of gene expression, which demonstrated that a subset of age-downregulated genes in the program are required for rapid proliferation of cultured fetal hepatocytes (see reference 2). The observed effects on cell proliferation in vitro were concordant with effects on body size reported in mice with targeted ablation of these genes and thus suggest that the previously observed altered body size represents a direct effect on cell proliferation, rather than only indirect effects such as systemic disease, placental dysfunction, or impaired prior organogenesis. Taken together, the findings imply that the multi-organ postnatal genetic program involves the downregulation of many genes, some of which are required for rapid proliferation in early life, supporting the hypothesis that part of this program contributes to growth deceleration. The finding that the program occurs concurrently in kidney, lung, heart, and liver provides a potential explanation for how growth slows coordinately in these organs, thus maintaining body proportions.
We next investigated the physiological mechanisms that drive the multi-organ postnatal genetic program. We showed that delaying growth by inducing tryptophan deficiency in juvenile rats caused a striking delay in the genetic program, suggesting that the multi-organ postnatal genetic program is not simply driven by a biological timing mechanism, but instead depends on growth (see reference 2). Thus, the program could, for example, be driven by increasing cumulative number of cell divisions undergone (a cell division counter) or by increasing tissue mass. The dependence of the program on growth is also supported by preliminary observations in hypothyroidism, another model of growth inhibition.
We also investigated the molecular mechanisms driving the growth-limiting genetic program and found that histone 3, lysine 4 trimethylation (H3K4me3) declined with age in multiple age-downregulated genes and multiple organs (see reference 2). Because H3K4me3 is a signature of permissive chromatin, the observed declining H3K4me3 may reflect the conversion of chromatin into a non-permissive state with body growth, which may thus orchestrate the observed downregulation of multiple genes.
Taken together, our findings support the following model to explain limitation of organ and body size in mammals. Somatic growth deceleration results from a multi-organ postnatal genetic program, primarily involving downregulation of a large set of growth-promoting genes. Because the growth-limiting genetic program occurs simultaneously in multiple tissues, the growth rate of various organs declines in a concerted fashion, which serves to maintain body proportions. The growth-limiting program depends not simply on age but on somatic growth itself and may be orchestrated by epigenetic mechanisms including declining H3K4me3. Therefore, growth leads to progression of the program with downregulation of many growth-promoting genes, which in turn causes growth of these organs to slow and eventually cease, thus setting a fundamental limit on adult organ size.
Spatial and temporal regulation of gene expression in the mammalian growth plate
In mammals, longitudinal bone growth occurs at the growth plates. These cartilaginous structures are organized into three distinct layers: the resting zone, the proliferative zone, and the hypertrophic zone. Growth plate chondrocytes undergo sequential differentiation from the resting to the proliferative to the hypertrophic state as their spatial position shifts.
To explore the mechanisms responsible for spatial regulation in the growth plate in an unbiased manner, we developed a microdissection method to separate postnatal rat growth plates into their constituent zones and then used microarray analysis to characterize the changes in gene expression that occur as chondrocytes undergo spatially associated differentiation (see reference 3). We confirmed our findings using real-time PCR and in situ hybridization. We then used bioinformatic approaches to identify functional pathways that may regulate these processes.
In the transition from the resting to the proliferative zone, the analysis implicated several functional pathways: VDR/RXR activation, PDGF signaling, BMP signaling, and notch signaling. The microarray analysis implicated other functional pathways in the transition from the proliferative to the hypertrophic zone: p53 signaling, cell-cycle G2/M regulation, cell-cycle G1/S regulation, ephrin receptor signaling, oncostatin M signaling, and BMP signaling. We also used the microarray findings to identify potential molecular markers for each zone of the growth plate. Using an empirical formula to rank genes based on their spatial expression pattern, we identified markers that show greater than 10-fold specificity for each of the growth plate zones. These markers of chondrocyte differentiation are likely to prove useful in future studies.
In addition to spatial regulation, the growth plate also undergoes important temporal regulation. Over time, proliferation slows in the growth plate, causing the rate of longitudinal bone growth to decrease and approach zero as the organism approaches adult size. The decline in proliferation is accompanied by gradual structural involution of the growth plate. To explore the underlying mechanisms, we used microdissected growth plates from 3-, 6-, 9-, and 12-week-old rats and analyzed gene expression using microarray (see reference 3). The analysis implicated several functional pathways in the developmental program of growth plate senescence: eicosanoid signaling, VDR/RXR activation, p38 MAPK signaling, and Wnt/beta-catenin signaling. We also identified molecular markers for growth plate senescence, which can now be used as indicators of the maturational state of the growth plate chondrocytes. Comparison of gene expression changes during chondrocyte differentiation and senescence did not reveal a substantial overlap, suggesting that the mechanisms that block proliferation as chondrocytes hypertrophy are distinct from those that restrict proliferation with increasing age.
We next explored the physiological process that drives programmed senescence of the growth plate. We previously reported that hypothyroidism in rats slowed both growth plate chondrocyte proliferation and growth plate senescence, suggesting that senescence is not dependent on age per se but rather on chondrocyte proliferation. However, one alternative explanation is that the observed slowing of growth plate senescence is a specific consequence of hypothyroidism. We reasoned that, if delayed senescence is a general consequence of growth inhibition, rather than a specific result of hypothyroidism, then senescence would also be slowed by other growth-inhibiting conditions. We therefore used tryptophan deficiency to temporarily inhibit growth in newborn rats for four weeks (see reference 4). We then allowed the animals to recover and studied the effects on growth plate senescence. We found that structural, functional, and molecular markers of growth plate senescence were delayed by prior tryptophan deficiency, indicating that the developmental program of senescence had occurred more slowly during the period of growth inhibition. Taken together with previous studies in hypothyroid rats, our findings support the hypothesis that delayed growth plate senescence is a general consequence of growth inhibition and hence that growth plate senescence is not simply a function of time per se but rather depends on growth.
Publications
- Finkielstain GP, Forcinito P, Lui JC, Barnes KM, Marino R, Makaroun S, Nguyen V, Lazarus JE, Nilsson O, Baron J. An extensive genetic program occurring during postnatal growth in multiple tissues. Endocrinology 2009 150:1791-1800.
- Lui JC, Forcinito P, Chang M, Chen W, Barnes KM, Baron J.Coordinated postnatal downregulation of multiple growth-promoting genes: evidence for a genetic program limiting organ growth. FASEB J 2010 24:3083-92.
- Lui JC, Andrade AC, Forcinito P, Hegde A, WeiPing Chen, Baron J, Nilsson O. Spatial and temporal regulation of gene expression in the mammalian growth plate. Bone 2010 46:1380-90.
- Forcinito P, Andrade AC, Finkielstain GP, Baron J, Nilsson O, Lui JC. Growth-Inhibiting Conditions Slow Growth Plate Senescence. J Endocrinol 2010, in press.
- Lui JC, Chen W, Barnes KM, Baron J. Changes in gene expression associated with aging commonly originate during juvenile growth. Mech Ageing Dev 2010, in press.
Collaborators
- Ola Nilsson, MD, PhD, Karolinska University Hospital, Stockholm, Sweden
- Sohyun Ahn, PhD, NICHD, NIH
- Weiping Chen, PhD, NIDDK, NIH
- Vasantha Padmanabhan, PhD, University of Michigan
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.