

You are here: Home > Section on Developmental Genetics
Heritable Neurodegenerative, Inflammatory, and Autoimmune Disorders

- Anil Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Zhongjian (Gary) Zhang, MD, PhD, Staff Scientist
- Sondra W. Levin, MD, Adjunct Scientist
- Chinmoy Sarkar, PhD, Visiting Fellow
- Goutam Chandra, PhD, Visiting Fellow
- Shiyong Peng, PhD, Visiting Fellow
- Eryan Kong, PhD, Visiting Fellow
- Ashleigh Bouchelion, BS, Medical Student
- Andrew Chen, Undergraduate Summer Intern
The Section on Developmental Genetics conducts laboratory and clinical investigations into neurodegenerative, inflammatory, and autoimmune disorders. Our research currently focuses on understanding the molecular mechanisms of pathogenesis of a group of hereditary childhood neurodegenerative lysosomal storage disorders called neuronal ceroid lipofuscinosis (NCL), commonly known as Batten disease. Mutations in eight genes cause various types of NCLs. Currently, there are no effective treatments for any of the NCLs. The infantile NCL or INCL is an autosomal recessive disease caused by mutations in the CLN1 gene, which encodes a lysosomal enzyme, palmitoyl-protein thioesterase-1 (PPT1). PPT1 catalyzes the cleavage of thioester linkage in palmitoylated (S-acylated) proteins (constituent of ceroid), facilitating their degradation in lysosomes. Thus, PPT1 deficiency causes accumulation of ceroid in lysosomes, leading to INCL pathogenesis. Children afflicted with INCL are normal at birth but, by 11 to 18 months of age, exhibit signs of psychomotor retardation. By 2 years of age, they are completely blind due to retinal degeneration and, by the age 4, manifest no brain activity and remain in a vegetative state for 6 to 8 years before eventual death. These grim outcomes underscore the urgent need for the development of rational and effective therapeutic strategies not only for INCL but also for all NCLs.
The aim of our clinical studies is to apply the knowledge gained from our laboratory investigations to develop novel therapeutic strategies for Batten disease. The results of our earlier investigations led to a bench-to-bedside clinical trial that is currently under way. Using CNL1-knockout (PPT1-KO) mice, which recapitulate virtually all clinical and pathological features of INCL, we discovered that PPT1 deficiency causes endoplasmic reticulum (ER) and oxidative stress, which at least in part causes neuronal death by apoptosis. We also delineated a mechanism by which PPT1 deficiency disrupts the recycling of the synaptic vesicle (SV) proteins, which are essential for regenerating fresh SVs to replenish the SV pool size at the nerve terminals in order to maintain uninterrupted neurotransmission. During the past year, we demonstrated that ER and oxidative stresses mediate neuronal apoptosis and neuroinflammation. Further, we showed that omega-3 and omega-6 fatty acids suppress ER and oxidative-stress in cultured neurons and neuronal progenitor cells from PPT1-KO mice, suggesting that these fatty acids may have therapeutic potential. We also discovered that nucleophilic small molecules with antioxidant properties ameliorate the neurological abnormalities in PPT1-KO mice and extend their lifespan. These and other related studies (uteroglobin, IgA nephropathy, etc.) provide insight into the complex mechanisms of heritable disorders of neurodegeneration, inflammation, and autoimmune diseases and identify several potential therapeutic targets. Our ongoing laboratory and clinical investigations have advanced our knowledge of these diseases. We plan to apply new findings from our laboratory to develop novel therapeutic approaches not only for INCL but also for other related diseases.
Disruption of the blood-brain barrier by T-helper 17 (TH17) lymphocytes is mediated via matrix metalloproteinases: amelioration by small molecules suppressing TH17-differentiation.
The disruption of the blood-brain barrier (BBB) is a serious complication in neurodegenerative disorders; however, the molecular mechanism(s) remains unclear, and the development of effective therapy remains challenging. Recent evidence indicates that CD4+ T-helper 17 (TH17) lymphocytes expressing the retinoic acid receptor–related orphan receptor γt (RORγt) cause BBB disruption and neuroinflammation. However, the mechanism by which these immune cells disrupt the BBB remains unknown. Using PPT1-KO mice, we tested the hypothesis that interleukin-17A (IL-17A) produced by TH17 lymphocytes stimulates production of matrix metalloproteinases (MMPs), which are known to degrade the tight-junction proteins between adjacent endothelial cells that are essential for maintaining BBB integrity. We found that, in PPT1-KO mice, the BBB disruption coincided with elevated levels of RORt (an orphan nuclear receptor)–positive TH17 lymphocytes and interleukin-17A (IL-17A). Moreover, the mice showed increased levels of MMPs. Furthermore, IL-17A-treatment of cultured brain endothelial cells (constituent of the BBB) stimulated production of MMPs. Importantly, dietary supplementation of resveratrol (RSV), an antioxidant/anti-inflammatory polyphenol, in PPT1-KO mice, which manifest oxidative stress and neuroinflammation, markedly reduced IL-17A and MMPs and elevated levels of tight-junction proteins. Intriguingly, RSV suppressed the differentiation of CD4+ T-lymphocytes to IL-17A–positive TH17 cells. We propose that, via MMPs, TH17 lymphocytes cause BBB disruption and suggest that anti-oxidative and anti-inflammatory small molecules that suppress TH17-differentiation are therapeutic targets for neurodegenerative disorders such as INCL.
Impairment of adaptive energy metabolism and increased levels of phosphorylated-S6 kinase-1 contribute to INCL pathogenesis and are ameliorated by resveratrol treatment.
Click image to enlarge.
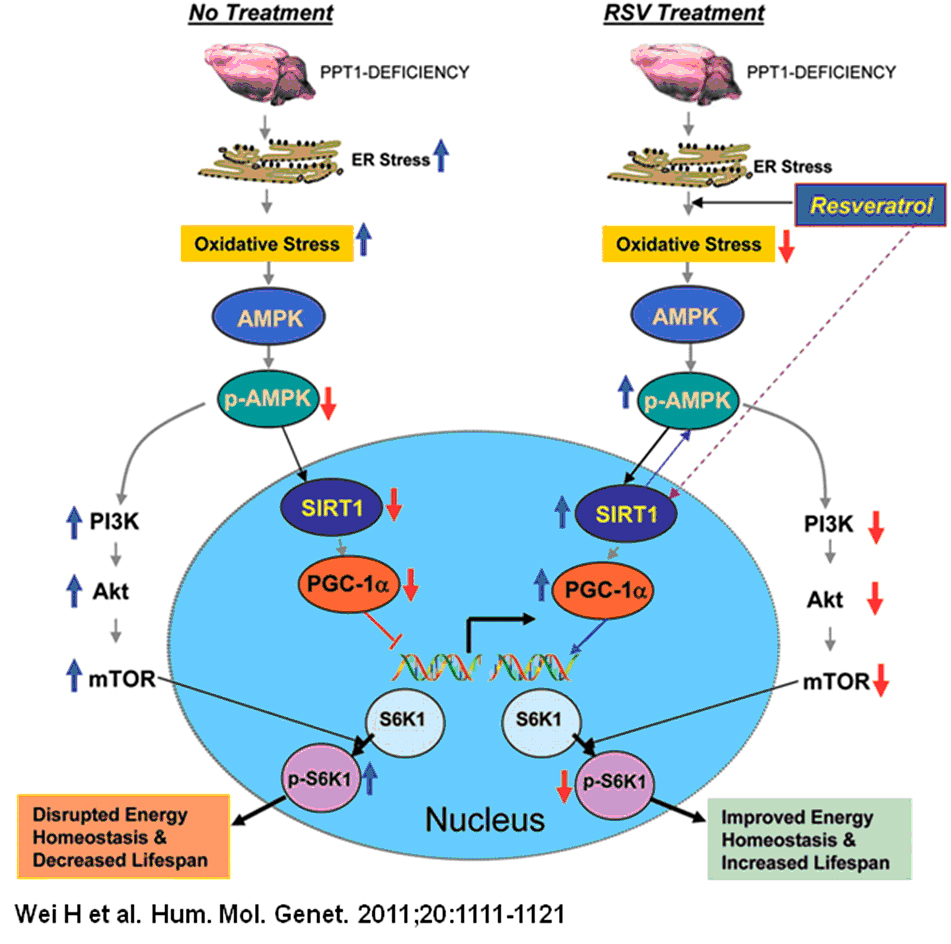
Disruption of adaptive energy metabolism is ameliorated by resveratrol.
A model explaining how resveratrol (RSV) may mediate energy homeostasis and lifespan expansion in Ppt1-KO mice. Increased oxidative stress and ER leads to neuronal apoptosis (upper left). Oxidative stress reduces levels of p-AMPK (phosphorylated AMPK) most likely by inhibiting the protein kinase LKB1, which is known to phosphorylate AMPK. Down-regulation of p-AMPK reduces the level and activation of SIRT1, which in turn reduces PGC-1alpha. As a result, expression of genes downstream of PGC-1alpha is down-regulated, disrupting energy homeostasis. Down-regulation of p-AMPK levels also stimulates PI3K (phosphatidylinositol 3-kinase), which in turn activates Akt. The activation of Akt leads to the phosphorylation and activation of mTOR (mammalian target of rapamycin), which is known to phosphorylate S6K1 (ribosomal S6 protein kinase 1). Increased p-S6K1 levels inversely correlate with lifespan. RSV, an SIRT1-activator and anti-oxidant polyphenol, not only reduces oxidative stress but also increases p-AMPK levels, most likely by stimulating LKB1. Thus, RSV stimulates the PGC-1alpha level, which increases the expression of genes downstream of PGC-1alpha. Activated SIRT1 may also enhance p-AMPK levels. By increasing p-AMPK levels, RSV also suppresses activation of the PI3K/Akt pathway of mTOR activation, which reduces p-S6K1 levels and thereby increases lifespan. Therefore, by stimulating the p-AMPK/SIRT1/PGC-1alpha signaling pathway and suppressing p-S6K1 levels, RSV improves energy homeostasis in neurons and in other cell types in PPT1-KO mice, causing a small increase in lifespan. As suggested by the results obtained from INCL fibroblasts, RSV may have similarly beneficial effects in INCL patients.
We previously reported that oxidative and ER stress, at least in part, mediate neuronal apoptosis in INCL as well as in PPT1-KO mice that mimic INCL. Given that mitochondria play critical roles in maintaining cellular energy homeostasis, we hypothesized that oxidative stress–mediated disruption of energy metabolism and homeostasis may contribute to INCL pathogenesis. In cultured INCL fibroblasts and in the brain tissues of PPT1-KO mice, we observed marked down-regulation of the NAD+/NADH ratio and levels of phosphorylated AMPK (p-AMPK), peroxisome proliferator-activated receptor-gamma (PPARgamma) coactivator-1alpha (PGC-1alpha), and Silent Information Regulator T1 (SIRT1), suggesting an abnormality in the AMPK/SIRT1/PGC-1alpha signaling pathway of energy metabolism. Moreover, we found that, in INCL fibroblasts and in the PPT1-KO mice, phosphorylated-S6K-1 (p-S6K1) levels, which inversely correlate with lifespan, are markedly elevated. Most important, resveratrol, an antioxidant polyphenol, elevated the NAD+/NADH ratio and levels of ATP, p-AMPK, PGC-1alpha, and SIRT1 while decreasing the level of p-S6K1 in both INCL fibroblasts and PPT1-KO mice, which showed a modest increase in lifespan. Our results indicate that oxidative stress–induced disruption of adaptive energy metabolism and increased levels of p-S6K1 are contributing factors in INCL pathogenesis and provide the proof of principle that small molecules such as resveratrol, which alleviate these abnormalities, may have therapeutic potential.
Oxidative stress–mediated apoptosis in the central nervous system of PPT1-KO mice is ameliorated by omega-3 and omega-6 fatty acids.
The nervous system is highly enriched with an enormous variety of complex lipids, among which polyunsaturated fatty acids (PUFAs) predominate. Despite their abundance in the mammalian brain, de novo synthesis of PUFAs such as arachidonic acid (AA) and docosahexanoic acid (DHA) does not occur. Thus, diet is the sole source of these fatty acids or their precursors. Epidemiological studies have suggested that increased consumption of the omega-3 (n-3) PUFA may reduce the risk of neurodegenerative disorders. Neuroprotective effects of PUFA have also been demonstrated, although a clear mechanism has not been defined. PUFA may also have protective effects against oxidative stress, which we have previously reported as a mediator of neuronal apoptosis in INCL and in the PPT1-KO mice. Moreover, emerging evidence indicates that oxygen free-radical damage to brain lipids, carbohydrates, proteins, and DNA may contribute to neuronal cell death, causing neurodegeneration. We hypothesized that PUFAs such as omega-3 and omega-6 fatty acids may have neuroprotective effects in PPT1-deficient cells. Accordingly, we analyzed cultured PPT1-deficient neurons and neuronal progenitor cells derived from the fetal brain tissues of PPT1-KO mice and explored the effects of PUFA treatment of these cells; we noted a marked reduction in the level of reactive oxygen species (ROS) and suppression of apoptosis. For the first time, our results demonstrate that while endogenous PUFA levels in the PPT1-KO mice appear to be similar to or even slightly higher than those of the wild-type littermates, PUFA treatment suppresses oxidative stress and reduces neuronal apoptosis induced by these stresses, raising the possibility of PUFAs' therapeutic potential for INCL.
Combination Cystagon™ and Mucomyst© treatment of patients with INCL: a bench-to-bedside protocol
Given that PPT1 catalyzes the cleavage of thioester linkages in palmitoylated (S-acylated) proteins (see above), its deficiency impairs degradation of these lipid-modified proteins by lysosomal proteases. Consequently, accumulation of these modified proteins (ceroids) in lysosomes leads to INCL pathogenesis. Given that thioester linkages are susceptible to nucleophilic attack, drugs that can cleave such linkages may have therapeutic potential for INCL. The results of our laboratory study showed that two drugs, phosphocysteamine and N-acetylcysteine, cleave thioester linkages in [14C] palmitoyl-CoA, a model substrate of PPT1, in lymphoblasts and fibroblasts from INCL patients. The drugs also facilitated the depletion of lysosomal ceroids and inhibited apoptosis. The results prompted us to initiate a bench-to-bedside clinical protocol to determine whether a combination of Cystagon™ (cysteamine bitartrate) and Mucomyst© (N-acetylcysteine) is beneficial for patients with INCL. Initially, the NICHD-IRB approved our protocol for the treatment of 20 INCL patients; the study has now been extended to include 40 INCL patients carrying any two mutations in the CNL1 gene. So far, we have treated 10 INCL patients; the study is currently recruiting patients and will continue until the approved number of patients is treated. The protocol admits INCL patients who are 6 months to 3 years of age. Patients may be referred to Dr. Mukherjee at mukherja@exchange.nih.gov.
Neurodegeneration in INCL mice evaluated by MRI and MR Spectroscopy
The effectiveness of potential INCL therapies in vivo could be evaluated by using PPT1-KO mice, which recapitulate virtually all clinical and pathological features of INCL. However, such studies require non-invasive methods to perform repeated evaluations on the same animal, which necessitates the development of such methods. We found that magnetic resonance imaging and magnetic resonance spectroscopy may be used to evaluate potential therapeutic agents on PPT1-KO mice to determine their effectiveness in vivo.
Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load, and suppresses apoptosis in cultured cells from INCL patients.
Nonsense mutations in the CNL1 gene generate premature termination codons, producing truncated, nonfunctional or deleterious proteins. PPT1 nonsense-mutations account for approximately 31% of INCL patients in the US. While aminoglycosides such as gentamycin suppress nonsense mutations, their inherent toxicity prohibits chronic use in patients. Given that PTC124 is a non-toxic compound that induces ribosomal read-through of premature termination codons, we sought to determine whether PTC124 treatment of cultured cells from INCL patients carrying nonsense mutations in the CNL1 gene would correct PPT1 enzyme deficiency with beneficial effects. Our results showed that PTC124 treatment of cultured cells from INCL patients carrying PPT1 nonsense mutations induced PPT1 enzymatic activity in a dose- and time-dependent manner. Even though PTC124 treatment of INCL cells achieved only a modest increase in PPT1 activity (virtually identical to that resulting from gentamycin treatment), the treatment reduced thioester (constituent of ceroid) load. Our results suggest that PTC124 treatment induces PPT1 enzymatic activity in cultured cells from INCL patients carrying PPT1 nonsense mutations and that such modest enzymatic activity has demonstrable beneficial effects on these cells. The clinical relevance of these effects may be tested in animal models of INCL carrying nonsense mutations in the CNL1 gene.

Gary Zhang, Anil Mukherjee, Eryan Kong, Sam Peng, Chinmoy Sarkar, and Goutam Chandra
Additional Funding
- Batten Disease Support and Research Association (BDSRA)
Publications
- Sarkar C, Zhang Z, Mukherjee AB. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol Genet Metab 2011;104:338-345.
- Wei H, Zhang Z, Saha A, Peng S, Chandra G, Quezado Z, Mukherjee AB. Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: partial rescue by resveratrol. Mum Mol Genet 2011;20:1111-1121.
- Mukherjee AB, Zhang Z. Genes, environment and allergic asthma. J Biol Chem 2011;286:32883-32889.
- Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, Mukherjee AB. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest 2008;118:3075-3086.
- Zhang Z, Butler JD, Levin SW, Wisniewski K, Brooks SS, Mukherjee AB. A novel approach towards treatment of infantile neuronal ceroid lipofuscinosis, a hereditary progressive encephalopathy of childhood. Nat Med 2001;7:478-484.
Collaborators
- Eva Baker, MD, PhD, Clinical Center, NIH, Bethesda, MD
- Andrea Gropman, MD, Children's National Medical Center Center for Neuroscience Research (CNR), Washington, D.C.
- Christian Hedberg, PhD, Max Planck Institute of Molecular Physiology, Dortmund, Germany
- Jeeva Munasinghe, PhD, NMR Center, NINDS, Bethesda, MD
- Zenaide Quezado, MD, Departments Anesthesiology and Pain Medicine, Children's National Medical Center, Washington, D.C.
- Satya P. Singh, PhD, Laboratory of Molecular Immunology, NIAID, Bethesda, MD
- Herbert Waldmann, PhD, Max Planck Institute of Molecular Physiology, Dortmund, Germany
- Yan Xu, PhD, Indiana University Medical School, Indianapolis, IN
Contact
For more information, email mukherja@exchange.nih.gov.