

You are here: Home > Section on Genetic Disorders of Drug Metabolism
Molecular Biology, Regulation, and Biochemistry of UDP-Glucuronosyltransferase Isozymes

- Ida S. Owens, PhD, Head, Section on Genetic Disorders of Drug Metabolism
- Nikhil K. Basu, PhD, Staff Scientist
- Amit Raychoudhuri, PhD, Visiting Fellow
- Sirsendu Jana, PhD, Visiting Fellow
- Mousumi Basu, BS, Technician Training Fellow
UDP-glucuronosyltransferase (UGT) isozymes, distributed primarily in liver, kidney, the gastrointestinal tract, and steroid-responsive tissues, carry out the essential function of converting innumerable and frequently encountered, but structurally diverse, lipophilic chemicals to glucuronides. Such lipophiles include toxic metabolites, dietary constituents, environmental agents/carcinogens, and therapeutics. Conversion of chemicals to glucuronides inactivates them and hastens excretion from the body to prevent tissue accumulation and toxicities. Neurotoxic bilirubin is the most important endogenous substrate, followed by genotoxic catechol estrogens. The UGT isozyme system prevents bilirubin neurotoxicities in children and inactivates common environmental mutagens and carcinogens but prematurely converts therapeutics. An understanding of the mechanism of glucuronidation would allow development of methods and strategies for accelerating removal of toxic chemicals while extending the therapeutic benefits of glucuronidatable medications. In addition to continuing to characterize the individual isozymes that constitute the human UGT chemical defense system, a major research aim of this laboratory has been to understand the basic feature(s)/mechanism(s) underlying the enormous range in substrate preference of each isozyme.
Expansion of the phosphorylation requirements of both UGT1A and 2B family members
Earlier findings from this laboratory include identification and characterization of: (i) the bilirubin-metabolizing UGT1A1 isozyme; (ii) the novel 215-kb UGT1A locus that encodes 13 independently regulated UGT genes including the UGT1A1 gene; and (iii) the first genetic defects in the UGT1A1 gene that lead to Crigler-Najjar diseases in children. We also cloned and identified the primary endogenous substrates for two critical UGT2B isozymes, UGT2B7 and UGT2B15. We continue to study both families of isozymes. Characterization of UGT2B7 demonstrated its essential role in detoxifying depurinating catechol estrogens that are associated with the initiation of breast cancer. We also discovered that UGT2B15 preferentially metabolizes dihydrotestosterone (DHT) and its primary metaboite 5alpha-androstane-3alpha,17beta-diol (3alpha-diol). According to UGT2B15's cellular distribution and biochemistry, the isozyme participates in DHT homeostasis and thus prevention of prostate cancer.
We demonstrated that UGTs require regulated phosphorylation, which can undergo reversible or irreversible downregulation, respectively, by dietary agents or by select kinase inhibitors. Detailed analysis of 5 out of 19 human UGTs demonstrated that regulated phosphorylation at their Ser/Thr and/or Tyr residues is reversibly inhibited by dietary curcumin. Our model studies with UGT1A7 mutated at PKC sites demonstrated that phosphorylation controls substrate selection, which changes in parallel with changes in pH catalysis optima. Co-immunoprecipitation studies of UGT1A7His and UGT1A10 revealed that PKCepsilon and PKCalpha/delta phosphorylate UGT1A7 and UGT1A10, respectively. Consistent with these observations, treatment of UGT1A7–transfected cells with the PKCepsilon–specific inhibitor peptide or general PKC inhibitors raised 17beta-estradiol catalysis 5- to 11-fold, with a parallel reduction in the level of phospho-serine-432. This novel mechanism involves PKC–mediated phosphorylation of UGT such that phospho-serine/threonine regulates substrate specificity in response to chemical exposures, possibly conferring survival benefit.
Substrate specificity of UGT2B7 is dictated by differential tyrosine kinase phosphorylation.
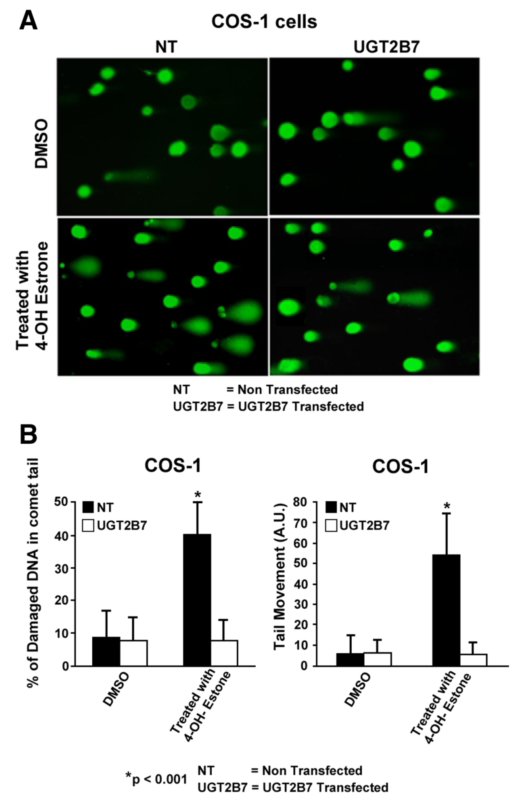
Click image to enlarge.
Figure 1. Effect of 2B7 transfection on 4-OHE1–damaged DNA in comet tails
A: Depiction of UGT2B7 protection against 4-OHE1–dependent depurination. B: Quantitation of protection against 4-OHE1–dependent depurination of COS-1 cells. The cells were treated with vehicle or 100 μm 4-OHE1 for 6 h, processed, and stained before evaluation of damaged DNA using Comet Score v1.5 software. The results for 200 cells/group were randomly and cumulatively evaluated from three experiments, using two different parameters as shown: percentage of damaged DNA in tail and tail moment (arbitrary units). Student's t test generated a p value of 0.001.
There is evidence that each UGT requires different phosphorylation for each isozyme. The UGT2B7 isozyme, which preferentially metabolizes genotoxic catechol estrogens, undergoes tyrosine phosphorylation at two distinct residues. Incorporation of immunoprecipitable [33P]orthophosphate into UGT2B7 following the enzyme's expression in COS-1 cells confirmed phosphorylation. The mutations Y236F-UGT2B7 and Y438F-UGT2B7 caused 90–100% loss of activity, indicating that tyrosine phosphorylation is required. Partial inhibition of activity by Src-specific PP2 with loss of anti–phospho-Y438-2B7 content, combined with similar disruption of co-localization and cross-linking of active c-Src and UGT2B7, strongly indicated that Src phosphorylates UGT2B7. Affinity-purified UGT2B7His, which was initially expressed in Src-free (SYF−/−) cells and phosphorylated in vitro by Src, confirmed that Src supports UGT2B7 phosphorylation based on both markedly enhanced anti–phospho-Y438-2B7 binding and activity. Unexpectedly, UGT2B7 expressed in SYF−/− and SYF+/− cells metabolized 4-OH-estrone (4-OHE1) at 10- and 3-fold higher rates, respectively, than when UGT2B7 was expressed in COS-1 cells; a similar analysis showed that 17beta-estradiol (E2) metabolism was 16- and 9-fold higher than in COS-1 cells. Anti-phospho-Y438-2B7 detected phospho-Y438-2B7 in each cell line, showing that unidentified tyrosine kinase(s) (TKs) phosphorylated UGT2B7 in SYF−/− cells. UGT2B7-transfected COS-1 cells treated with the Src-specific inhibitor PP2 exhibited down-regulated 4-OHE1 glucuronidation with simultaneously up-regulated E2 metabolism. This finding indicates that PP2 inhibition of Src allows E2 metabolism owing to UGT2B7 phosphorylation by unidentified TK(s). Importantly, UGT2B7 expressed in SYF−/− is more competent at metabolizing E2 in cellulo than UGT2B7 expressed in COS-1 cells. To confirm that Src-controlled UGT2B7 prevents toxicity, we showed that UGT2B7 transfected into COS-1 cells efficiently protected against 4-OH-E1–mediated depurination. Our results reveal that Src-dependent phosphorylation of UGT2B7 allows metabolism of 4-OHE1, but not of E2, in COS-1 cells, while non-Src–phosphorylated UGT2B7 metabolizes both chemicals. Importantly, we determined that UGT2B7 substrate selection is not fixed but varies according to the TK(s) carrying out phosphorylation. Lastly, UGT2B7 transfection of COS-1 cells provided greater than 80% protection against 4-OH-estrone−based depurination. While c-Src supports the required and regulated phosphorylation of UGT2B7, which is disrupted in breast carcinomas, functional UGT2B7 protects against catechol estrogen depurination, which is linked to tumor initiation. This study showed that UGT2B7, like UGT1A7 (Basu et al., Proc Natl Acad Sci USA 2005;102, 6285), uses phosphorylation of tyrosine residues with different tyrosine kinases to control substrate specificity (4).
UGT2B15 and UGT2B17 dependence on PKC phosphorylation with separate regulation by Src kinase phosphorylation
With regard to homeostasis of dihydrotestosterone (DHT) in prostate tissue, both UGT2B15 and UGT2B17, distributed in prostate luminal and basal cells, respectively, glucuronidate the hormone and its 3alpha-diol metabolite to facilitate their clearance from the prostate. Both UGT2B15 and UGT2B17 have a strict requirement for regulated PKC phosphorylation at position S172 and a complex pattern of phosphate regulation at position Y237, evidently via Src. Analyses also show that UGT2B15 activity is modified by phosphorylation at three other sites: Y99, S124, and S422. The Y99 and S422 sites are also present in UGT2B17, but mutations at the sites have no effect on activity. Co-immunoprecipitation studies show that PKC-alpha binds to UGT2B15 while PKC-alpha and epsilon bind to UGT2B17. Lastly, UGT2B15 expressed in Src-free cells undergoes proteasomal ubiquinationation, which is inhibited by the specific inhibitor MG132, indicating that UGT2B15 not only requires Src for its phosphorylation but also for its maintenance. In contrast, the activity of UGT2B17 expressed in Src-containing COS-1 cells increases two-fold following Src-siRNA treatment or following expression in Src-free cells, indicating that UGT2B17's activity is reduced in the presence of Src. Hence, Src has opposite effects on UGT2B15 and UGT2B17. The fact that Src has opposite effects on luminal cell–distributed UGT2B15 and basal cell–distributed UGT2B17, with the two cell types juxtaposed in a stratified manner, suggests that the arrangement accomodates the exchange of small molecules via jap-junction structures between the cells. It is well known that luminal cells carry out all prostate-specific functions, which are dependent on the DHT–occupied androgen receptor. While the function of basal cells is not well understood, it has been suggested the basal cell supports the luminal cell in carrying out prostate-specific functions. Hence the inverse effects of Src suggest that the 25- to 50-fold greater activity of UGT2B17 in basal cells than of UGT2B15 in luminal cells is a rationale for the structure of the prostate and the biochemistry of DHT and that intercellular communications are thus important for regulating DHT levels in luminal cells. The model proposes that testosterone produced in basal cells travels via gap-junctions to luminal cells for conversion to DHT and that excess androgen is transported back to basal cells for glucuronidation by robust UGT2B17 activity.
UGT phosphorylation is requlated via signaling.
Catalase or herbimycin-A inhibition of constitutive or hydrogen peroxide–activated UGTs implicated ROS-related oxidants as second messengers in maintaining constitutive PKC–dependent signaling—evidently sustaining UGT phosphorylation and activity. Given that cells use signal transduction to detect and respond appropriately to environmental changes, these findings, combined with our earlier demonstration that specific phospho-groups in UGT1A7 determined substrate selections, suggest that regulated phosphorylation allows adaptations regarding differential phosphate utilization by UGTs for the isozymes to function efficiently.
Tertiary structure of UGT(s)
To investigate further the requirements of phosphorylation of ER–bound UGTs, we attempted to purify a catalytically active UGT protein for structural analysis. UGT1A7- and UGT2B7-cDNAs, adapted with thrombin/his/myc affinity ligands, not only establish a highly effective UGT–solubilizing system that retains activity but also enabled us to isolate UGT–containing complexes involved in PKC and/or tyrosine kinase signaling pathways similar to those described for other cellular processes. The complex contained one 58 kDa UGT1A7His and one 110 kDa beta-COP for every two 29 kDa 14-3-3 phospho-serine chaperone proteins. We found that, for PKC–dependent signaling, UGT1A7 associated with RACKepsilon in a 225-kDa adapter complex that included the phosphoserine-dependent 14-3-3 protein. Mutation of UGT1A7His at its 14-3-3 binding sites led to marked instability of solubilized UGT1A7His. Like all UGTs, UGT1A7 has two 14-3-3 binding sites: S162 and T403. Mutations at these sites indicate that 14-3-3 also stabilizes UGT stored at 4°C. These important findings should enable us to isolate a UGT and carry out analysis of its tertiary structure as well as identify events and components involved in phosphorylation-dependent signaling. It is notable that 14-3-3 binding sites exist in all UGTs. We confirmed binding of UGTs to phosphoserine–14-3-3 by co-immunoprecipitation and/or co-localization with UGT1A1His, UGT1A6His, UGT1A7His, UGT1A10, or UGT2B7His. In summary, our studies support the notion that UGT1A7 exists as a cellular complex(es) that sustains activity via protein kinase(s) signaling. Our findings lay the foundation for further studies concerning this critical endogenous chemical-defense enzyme system.
Structural analysis and identification of the common donor-substrate binding site in UGT1A10
Because of the difficulties associated with purifying ER–bound UGTs for structural studies, we carried out homology-based computer modeling to aid analysis. The search found structural homology in Escherichia coli UDP-galactose 4-epimerase. Consistent with predicted similarities involving the common UDP moiety in substrates, Lineweaver-Burke plots showed that UDP-glucose and UDP-hexanol caused competitive inhibition. Among the predicted binding sites N292, K314, K315, and K404 in UGT1A10, we found two informative sets of mutants: the K314R/Q/A/E/G mutants had null activities; that of K404R was 2.7-fold higher than WT; and K314/E had 50% less activity. Scatchard analysis of binding of the affinity-ligand 5-azido-uridine-[beta-32P]-diphosphoglucuronic acid to purified UGT1A10-His or UGT1A7-His revealed high- and low-affinity binding sites. UGT1A10-His bound to radiolabeled affinity ligand and digested with 2-nitro-5-thiocyanobenzoic acid (NTCB) revealed an 11.3-kDa and a 14.3-kDa peptide associated with K314 and K404, respectively. Similar treatment of UGT1A10His-K314A bound to the ligand lacked both peptides; UGT1A10-HisK404R and UGT1A10-HisK404E showed 1.3-fold greater and 50% less label in the 14.3-kDa peptide, respectively, compared with UGT1A10-His, without affecting the 11.3-kDa peptide. Scatchard analysis of binding data of the affinity ligand to UGT1A10His-K404R and UGT1A10His-K404E showed a six-fold reduction and a large increase in Kd, respectively. Our results indicate that K314 and K404 are required UDP-glcA binding sites in UGT1A10, that K404 controls activity and high affinity sites, and that K314 and K404 are strictly conserved in 70 aligned UGTs, except for S321—equivalent to K314—in UGT2B15 and UGT2B17 and I321 in the inactive UGT8, which suggests that UGT2B15 and UGT2B17 have suboptimal activity. Our data thus strongly support UDPglcA binding to K314 and K404 in UGT1A10.
Publications
- Basu NK, Kole L, Basu M, Chakraborty K, Mitra PS, Owens IS. The major chemical detoxifying system of UDP-glucuronosyltransferases requires regulated phosphorylation supported by protein kinase C. J Biol Chem 2008;283:23048-23061.
- Banerjee R, Pennington MW, Garza A, Owens IS. Mapping the UDP glucuronic acid binding site in UDP-glucuronosyltransferase-1A10 by homology-based modeling: confirmation with biochemical evidence. Biochemistry 2008;47:7385-7392.
- Mitra PS, Basu NK, Owens IS. Src supports UDP-glucuronosyltransferase-2B7 detoxification of catechol estrogens associated with breast cancer. Biochem Biophys Res Commun 2009;382:651-656.
- Mitra PS, Basu NK, Basu M, Chakraborty S, SahaT, Owens IS. Regulated phosphorylation of a major UDP-glucuronosyltransferase isozyme by tyrosine kinases dictates endogenous substrate selection for detoxification. J Biol Chem 2011;286:1639-1648.
Collaborators
- Antony McDonagh, PhD, University of California San Francisco, San Francisco, CA
- Masahiko Negishi, PhD, Laboratory of Reproductive and Developmental Toxicology, NIEHS, Research Triangle Park, NC
- Juan Rivera, PhD, Molecular Immunology and Inflammation Branch, NIAMS, Bethesda, MD
- Tapas Saha, PhD, Lombardi Comprehensive Cancer Center, Georgetown University, Washington, D.C.
Contact
For further information, contact ida.owens2@nih.gov.