

You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Postdoctoral Fellow
- Sao Fong Cheung, Predoctoral Fellow
- Angela Delaney, MD, Clinical Fellow
- Fariha Kamran, MD, Clinical Fellow
- Youn Hee Jee, MD, Clinical Fellow
- Stephan S. Spath, PhD, Special Volunteer
We investigate the cellular and molecular mechanisms governing childhood growth and development. We are especially interested in mechanisms that allow rapid cell proliferation and hence rapid body growth in young mammals and that subsequently suppress proliferation, thus setting a fundamental limit on the adult body size of the species. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. In addition, further investigation of the identified growth-limiting mechanisms may lead to broader medical applications, because disruption of these mechanisms may contribute to oncogenesis, and conversely transient therapeutic suspension of growth-limiting mechanisms in adult cells might be used to achieve tissue regeneration.
Growth limitation and aging
Aging involves a gradual decline in physiological function throughout the organism, decreasing the ability to respond to stress and homeostatic imbalance, and increasing susceptibility to disease. The causes of aging are poorly understood. One mechanism that may contribute to aging is a progressive decline in the proliferative capacity of various cell types, including adult stem cells, which impairs tissue maintenance and regenerative potential. For example, with age, there is a declining ability of the liver to regenerate after partial hepatectomy.
Declining proliferative capacity in many tissues does not begin during adult life but initiates much earlier. In embryonic and early postnatal life, many tissues show rapid replication, leading to rapid somatic growth. Subsequently, cellular mechanisms slow this robust proliferation, such that, by adulthood, most tissues have entered a quiescent state in which proliferation occurs only as needed to replace dying cells. We recently found evidence that the mechanism responsible for this decline in juvenile cell proliferation involves a genetic program that is common to many organs and includes the downregulation of multiple growth-promoting genes with age.
We hypothesized that the mechanisms that progressively restrain juvenile growth continue to progress into adult life, thus contributing to the decline in proliferative capacity associated with aging. Progression of the underlying proliferation-limiting genetic program might lead first to cellular quiescence in young adulthood, such that some cells can still proliferate when stimulated for tissue renewal, but then, during aging, continued progression of the program might further limit the proliferative capacity of cells to a level below that needed for tissue renewal.
As an initial test of this hypothesis, we used expression microarray to compare changes in gene expression that occur in liver, kidney, and lung of mice during adult aging with changes that occur in juvenile life, as somatic growth slows. We found that many of the changes in gene expression that occur during adult aging originate in early postnatal life, during the juvenile period of growth deceleration (Figure 1). Furthermore, bioinformatic analysis of the genes that showed persistent changes in expression, both during juvenile life and during adult aging, indicate that cell-cycle related genes are strongly over-represented.
Thus, the findings support the hypothesis that the genetic program that slows growth in juvenile life in order to limit adult body size persists into adulthood, and may eventually hamper maintenance and repair of multiple organs and thus contribute to the aging process. This hypothesis might provide an explanation for the observation that small mammals generally undergo both aging and suppression of juvenile growth on a far shorter time scale than do large mammals. The hypothesis might also help explain why growth-inhibiting conditions such as caloric restriction, growth hormone deficiency, or insulin-like growth factor-1 deficiency slow aging. We have previously shown that growth-inhibiting conditions slow the juvenile growth-limiting genetic program. This slowing of the program may then conserve proliferative capacity and therefore slow aging.
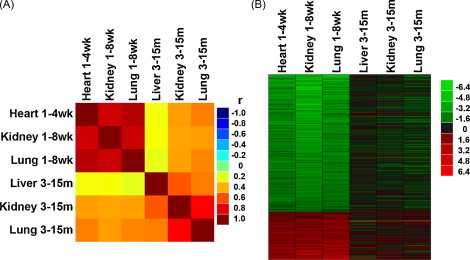
Figure 1. Changes in gene expression occurring during postnatal growth deceleration continued during aging.
The analysis involved 316 genes that showed coordinate changes in genes expression in multiple organs of mice and rats during juvenile growth. (A) Correlation analysis was performed between temporal changes in gene expression during juvenile growth (1 week vs. 8 weeks) and aging (3 months vs. 15 months) in various organs of mice. The correlation coefficients (r values) are depicted in a color map. Yellow-red, positive correlation; green, no correlation (r = 0); cyan-blue, negative correlation. (B) Heat maps were constructed from microarray data for the same set of 316 genes. Green represents downregulated genes, and red represents upregulated genes compared with the baseline (3 months for aging, 1 week for postnatal growth). The color intensity corresponds to the magnitude of the change from baseline (log2 [value at later time point/value at baseline time point]). For genes that were downregulated from 1 to 8 weeks, there was a strong tendency for continued decline during aging. Similarly, genes upregulated during juvenile life tended to continue that upregulation during aging.
Mechanisms limiting skeletal growth
Mammalian body length is primarily determined by bone elongation, which occurs at the growth plate. These cartilaginous structures are organized into three distinct layers—the resting zone, the proliferative zone, and the hypertrophic zone. Growth plate chondrocytes undergo sequential differentiation from the resting to the proliferative to the hypertrophic state as their spatial position shifts.
Bone elongation is rapid in early life but gradually slows with age, until growth velocity eventually approaches zero in adulthood. The decline in longitudinal bone growth with age is associated with functional, structural, and molecular changes in the growth plate. These senescent changes include a decline in the chondrocyte proliferation rate, the overall growth plate height, proliferative, and hypertrophic zone heights, and in column density and extensive changes in gene expression.
Based on prior findings, we hypothesized that growth plate senescence is not simply dependent on time per se but rather depends on chondrocyte proliferation. Thus, for example, growth plate chondrocytes may have a finite proliferative capacity, which is gradually exhausted, leading to a decline in growth rate and other senescent changes in the growth plate.
To test this hypothesis, we used a tryptophan-deficient diet to suppress growth temporarily in newborn rats. Afterwards, growth in these rats was allowed to recover by switching to a replete diet. We found that structural, functional, and molecular markers of growth plate senescence were delayed by prior tryptophan deficiency, indicating that the developmental program of senescence had occurred more slowly during the period of growth inhibition.
Combined with previous findings using other models of growth inhibition, the cumulative evidence supports the hypothesis that delayed growth plate senescence is a consequence of growth inhibition, which in turn suggests that growth plate senescence is not simply a function of time per se but rather depends on growth. We have also found evidence that growth deceleration in non-skeletal tissues is similarly driven by growth itself, indicating that growth in many tissues is fundamentally limited by a negative feedback loop.
Regulation of skeletal growth by Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP)

Click image to enlarge.
Figure 2. LacZ expression in proximal tibiae of Gli1-lacZ mice at E16, 1-, 4-, 8, and 12 weeks of age
These mice have one allele in which coding sequences of Gli1 were replaced with lacZ, which encodes a β-galactosidase enzyme. Because hedgehog signaling stimulates Gli1 transcription, lacZ expression in this mouse indicates activity of the hedgehog pathway. Frozen sections (10 μm) of whole tibia and growth plate were stained for β-galactosidase activity using X-gal, a chromogenic substrate that yields a blue product. Representative photomicrographs are shown. The left, middle, and right hand columns show progressively increasing magnification, with the right hand column images centered on the periarticular cartilage. The lowest row shows a 1-week-old wild-type animal that served as a negative control. In general, lacZ expression was detected throughout the growth plate but at lower levels in hypertrophic cartilage. A, articular surface; RP, round proliferating chondrocytes; FP, flat proliferating chondrocytes; R, resting zone; P, proliferative zone; H, hypertrophic zone; PC, perichondrium; O, secondary center of ossification; wt, wild type.
Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP) are secreted proteins that act as paracrine signals, participating in a negative feedback loop that regulates chondrocyte differentiation and proliferation. However, the role of these proteins has primarily been studied in embryonic growth cartilage. Postnatally, this region undergoes major structural and functional changes.
To explore the organization of the Ihh-PTHrP system in the postnatal growth plate, we microdissected growth plates of 7-day-old rats into their constituent zones and assessed expression of genes participating in the Ihh-PTHrP feedback loop. Ihh, Ptch1, Smo, Gli1, Gli2, Gli3, and Pthr1 were expressed in regions analogous to the expression domains in embryonic growth cartilage. PTHrP, however, was expressed in resting zone cartilage, a site that differs from the embryonic source, the periarticular cells. We then used mice in which lacZ has replaced coding sequences of Gli1 and thus serves as a marker for active hedgehog signaling. At 1-, 4-, 8-, and 12-weeks of age, lacZ expression was detected in a pattern analogous to that of embryonic cartilage (Figure 2).
The findings support the hypothesis that the embryonic Ihh-PTHrP feedback loop is maintained in the postnatal growth plate except that the source of PTHrP has shifted from its embryonic location in the periarticular cartilage to a more proximal location in the resting zone.
Publications
- Forcinito P, Andrade AC, Finkielstain GP, Baron J, Nilsson O, Lui JC. Growth-inhibiting conditions slow growth plate senescence. J Endocrinol 2010;208:59-67.
- Lui JC, Chen W, Barnes KM, Baron J. Changes in gene expression associated with aging commonly originate during juvenile growth. Mech Ageing Dev 2010;131:641-9.
- Chau M, Forcinito P, Andrade AC, Hegde A, Ahn S, Lui JC, Baron J, Nilsson O. Organization of the Indian hedgehog - parathyroid hormone-related protein system in the postnatal growth plate. J Mol Endocrinol 2011;47:99-107.
- Lui JC, Baron J. Mechanisms limiting body growth in mammals. Endocr Rev 2011;32:422-40.
- Lui C, Nilsson LO, Baron J. Growth plate senescence and catch-up growth. Endocr Dev 2011;21:23-9.
Collaborators
- Ola Nilsson, MD, PhD, Karolinska University Hospital, Stockholm, Sweden
- Vasantha Padmanabhan, PhD, University of Michigan, Ann Arbor, MI
- Sohyun Ahn, PhD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.