

You are here: Home > Section on Molecular Dysmorphology
Cholesterol Metabolism and Genetic Syndromes

- Forbes D. Porter, MD, PhD, Head, Section on Molecular Dysmorphology
- Chris A. Wassif, MSc, Technical Specialist
- Stephanie Cologna, PhD, Postdoctoral Fellow
- Celine Cluzeau, PhD, Postdoctoral Fellow
- Nicole M. Yanjanin, CPNP, Nurse Practitioner
- Sandra K. Conley, CPNP, Nurse Practitioner
- Cynthia Toth, BS, Technical Intramural Research Training Awardee
- Rao Fu, BA, Doctoral Student
- Elexa Ely, BS, Postbaccalaureate Fellow
- Michelle Dail, BA, Postbaccalaureate Fellow
- Stephan Siebel, MD, Clinical Fellow
- Elaine Tierney, MD, Special Volunteer
- Andrea Silvis, RN, Postbaccalaureate Fellow
- Ryan Lee, MD, Special Volunteer
- Simona Bianconi, MD, Special Volunteer
We study the molecular, biochemical, and cellular processes that underlie genetic disorders resulting from to impaired cholesterol homeostasis. The disorders include malformation/cognitive impairment syndromes due to inborn errors of cholesterol synthesis, neurodegenerative disorders due to impaired intracellular cholesterol and lipid transport, and Autistic Spectrum disorder (ASD) associated with low cholesterol. Human malformation syndromes resulting from inborn errors of cholesterol synthesis include Smith-Lemli-Opitz syndrome (SLOS), lathosterolosis, desmosterolosis, X-linked dominant chondrodysplasia punctata type 2 (CDPX2), and the CHILD syndrome. Neimann-Pick disease type C (NPC) results from impaired intracellular transport of cholesterol and lipids leading to neuronal loss. Our basic research uses mouse models of these genetic disorders to understand the biochemical, molecular, cellular, and developmental processes that underlie the birth defects and clinical problems encountered in affected patients. Our clinical research focuses on translating basic findings to the clinic. Natural history trials of both SLOS and NPC are ongoing. Our emphasis on both basic and clinical research allows us to integrate laboratory and clinical data in order to increase our understanding of the pathological mechanisms underlying both SLOS and NPC, with the goal of improving clinical care of these patients. Therapeutic trials have been conducted for both disorders, and future trials are being organized.
Smith-Lemli-Opitz syndrome and hyopcholesterolemic Autism Spectrum disorder
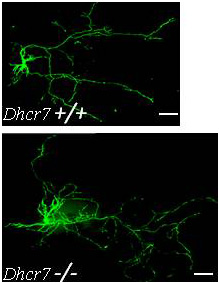
Figure 1. Increased dendrite and axon elongation of Dhcr7-mutant hippocampal neurons
Hippocampal neurons from Dhcr7 control and mutant E18.5 embryos were transfected with green flourescent protein after seven days and imaged three days later. Both the axon and dendrites from Dhcr7-mutant neurons were significantly longer than those of control neurons. This finding is consistent with dysregulated Rho GTPase activity, which we described in our mouse model of Smith-Lemli-Opitz syndrome; such dysregulation may contribute to the cognitive impairment associated with this inborn error of cholesterol synthesis.
SLOS is an autosomal recessive, multiple-malformation syndrome characterized by dysmorphic facial features, cognitive impairment, hypotonia, poor growth, and variable structural anomalies of the heart, lungs, brain, limbs, gastrointestinal tract, and genitalia. The SLOS phenotype is extremely variable. At the severe end of the phenotypic spectrum, infants often die as result of multiple major malformations while at the mild SLOS combines minor physical malformations with behavioral and learning problems. The syndrome is attributable to an inborn error of cholesterol biosynthesis that blocks the conversion of 7-dehydrocholesterol (7-DHC) to cholesterol.
Our laboratory initially cloned the human 3beta-hydroxysterol delta7-reductase gene (DHCR7) and demonstrated mutations of the gene in SLOS patients. Together with others, we have identified over 100 mutations of DHCR7 to date. We have also used gene targeting in murine embryonic stem cells to procuce multiple SLOS mouse models, including a null deletion and a hypomorphic point mutation. Mouse pups homozygous for the null mutation (Dhcr7Δ3-5/Δ3-5) exhibit variable craniofacial anomalies, are growth-retarded, feed poorly, and appear weak. Dhcr7Δ3-5/Δ3-5 pups die during the first day of life because they fail to feed; thus, we were not able use them for studying postnatal brain development, myelination, or behavior or to test therapeutic interventions. For this reason, we developed a mis-sense allele (Dhcr7T93M). The T93M mutation is the second most common mutation found in SLOS patients. Dhcr7T93M/T93M and Dhcr7T93M/Δ3-5 mice are viable and demonstrate SLOS with a gradient of biochemical severity (Dhcr7 Δ3-5/Δ3-5>Dhcr7 T93M/Δ3-5>Dhcr7T93M/T93M). We used Dhcr7T93M/Δ3-5 mice to test the efficacy of therapeutic interventions on tissue sterol profiles. As expected, dietary cholesterol therapy improved the sterol composition in peripheral tissues but not in the central nervous system. Treatment of mice with simvastatin improved the biochemical defect in both peripheral and central nervous system tissue, suggesting that simvastatin therapy may be used to treat some of the behavioral and learning problems in children with SLOS.
As part of our clinical studies on SLOS, we identified a novel oxysterol, 27-hydroxy-7-dehydrocholesterol (27-7DHC), derived from 7-DHC in SLOS patients. We therefore investigated whether 27-7DHC contributes to the pathology of SLOS and found a strong negative correlation between plasma 27-7DHC and cholesterol levels in SLOS patients. In addition, previous work showed that low cholesterol levels impair hedgehog signaling. Therefore, we hypothesized that increased 27-7DHC levels would have detrimental effects during development in response to suppression of cholesterol levels. To test our hypothesis, we produced SLOS mice (Dhcr7 Δ3-5/Δ3-5) expressing a CYP27 transgene. CYP27Tg mice have elevated CYP27 expression and 27-hydroxycholesterol levels but normal cholesterol levels. Dhcr7 Δ3-5/Δ3-5 mice are growth-retarded, exhibit a low incidence of cleft palate (9%), and die during the first day of life. Dhcr7Δ3-5/Δ3-5:CYP27Tg embryos are stillborn and have multiple malformations, including growth retardation, micrognathia, cleft palate (77%), lingual and dental hypoplasia, ankyloglossia, umbilical hernia, cardiac defects, cloacae, curled tails, and limb defects. We observed autopod defects (polydactyly, syndactyly, and oligodactyly) in 77% of the mice. Consistent with our hypothesis, sterol levels were halved in the liver and 20-fold lower in the brain tissue of Dhcr7 Δ3-5/Δ3-5 :CYP27Tg than in Dhcr7 Δ3-5/Δ3-5 embryos. The fact that 27-7DHC plays a role in SLOS may explain some of the phenotypic variability and may lead to development of a therapeutic intervention. This project is a prime example of how our group integrates both clinical and basic science to both understand the pathology of SLOS and develop potential therapeutic interventions.
To gain insight into pathophysiological processes contributing to SLOS, we completed a series of proteomic experiments to identify proteins that are differentially expressed in the cortex of Dhcr7Δ3-5/Δ3-5 embryos. Functional analysis is in progress; however, we have already identified alterations in dendrite and axon formation that may contribute to the cognitive deficits found in SLOS. In collaboration with investigators at Children's Hosptital Oakland Research Institute, we are using our hypomorphic mouse model to investigate adeno-associated viral gene therapy for SLOS.
We continue to use the SLOS mouse models to understand the pathophysiological processes that underlie the birth defects and clinical problems in SLOS and are applying various biochemical, molecular, and proteomic approaches to investigate these issues. We are also using the mouse models to test various prenatal and postnatal therapeutic interventions.
We are involved in an ongoing Natural History trial that is part of a NICHD/ORD–supported Rare Disease Research Consortium. Given that SLOS patients have a cholesterol deficiency, they may be treated with dietary cholesterol supplementation. To date, we have evaluated over 75 SLOS patients and continue to follow many of them in a longitudinal natural history study. We have just recently completed a randomized, placebo-controlled, double-blinded cross-over trial to study the safety and efficacy of simvastatin therapy in SLOS. As part of a Bench-to-Bedside proposal, we continue, in collaboration with Fran Platt's laboratory, to study impaired cholesterol and glycosphingolipid transport in SLOS and to investigate novel therapeutic interventions.
One reason for studying rare genetic disorders is to gain insight into more common disorders. Most patients with SLOS exhibit autistic characteristics. We are currently collaborating with groups from both NHGRI and the Kennedy Krieger Institute in Baltimore to analyze this association further. We received funding from Autism Speaks to initiate a multicenter double-masked placebo-controlled trial of cholesterol in hypocholesterolemic ASD. We also recently received funding from the NIH Director's Office to support whole-exome sequencing studies aimed at elucidating the genetic mechanism underlying hypocholesterolemia in ASD patients.
Lathosterolosis, desmosterolosis, and HEM dysplasia
Lathosterol 5-desaturase catalyzes the conversion of lathosterol to 7-dehydrocholesterol, representing the enzymatic step immediately preceding the defect in SLOS. Thus, to understand further the roles of reduced cholesterol versus elevated 7-dehydrocholesterol in SLOS, we disrupted the mouse lathosterol 5-desaturase gene (Sc5d) by using targeted homologous recombination in embryonic stem cells. Sc5d −/− pups are stillborn, present with micrognathia and cleft palate, and exhibit limb-patterning defects. Many of the malformations in these mutant mice resemble malformations in SLOS and are consistent with impaired hedgehog signaling during development. Biochemically, the mice exhibit markedly elevated lathosterol levels and reduced cholesterol levels in serum and tissue.
One goal of producing a lathosterolosis mouse model was to gain phenotypic insight for the purpose of identifying a corresponding human malformation syndrome. We have now identified a human infant patient with lathosterolosis, a human malformation syndrome not yet described. Biochemically, fibroblasts from the patient show reduced cholesterol and elevated lathosterol levels. Mutation analysis showed that the patient is homozygous for a single A→C nucleotide change at position 137 in SC5D, resulting in a mutant enzyme in which the amino acid serine is substituted for tyrosine at position 46. Both parents are heterozygous for the mutation. The infant's phenotype resembled severe SLOS. Malformations found in both the human patient and mouse model include growth failure, abnormal nasal structure, abnormal palate, micrognathia, and postaxial polydactyly. A unique feature of lathosterolosis is the clinical finding of mucolipidosis in the affected infant, which is not reported in SLOS and may help distinguish SLOS clinically from lathosterolosis. A lysosomal storage disorder, lathosterolosis may be replicated in embryonic fibroblasts from the Sc5d mutant mouse model. To distinguish pathological changes attributable to reduced cholesterol from those that are a consequence of elevated 7-DHC in SLOS, we are comparing proteomic changes in the Sc5d mutant mouse model with those in the SLOS mouse model.
Desmosterolosis is another inborn error of cholesterol synthesis that resembles SLOS. It results from a mutation in the 3beta-hydroxysterol delta24-reductase gene (DHCR24). DHCR24 catalyzes the reduction of desmosterol to cholesterol. We disrupted the mouse Dhcr24 gene by using targeted homologous recombination in embryonic stem cells. Surprisingly, although most Dhcr24 mutant mice die at birth, the pups are phenotypically normal.
Others have shown that mutations of the lamin B receptor (LBR) cause HEM (hydrops, ectopic calcification, moth-eaten skeletal) dysplasia in humans and ichthyosis in mice. LBR has both lamin B–binding and sterol Δ14-reductase domains. Although only a minor sterol abnormality has been reported, it has been proposed that LBR is the primary sterol Δ14-reductase and that impaired sterol Δ14-reduction underlies HEM dysplasia. However, DHCR14 also encodes a sterol Δ14-reductase. To test the hypothesis that LBR and DHCR14 are redundant sterol Δ14-reductases, we obtained ichthyosis mice (LbrSc5d−/−) and disrupted Dhcr14. Dhcr14 Sc5d−/− mice are phenotypically normal. We found no sterol abnormalities in either Lbr Sc5d−/− or Dhcr14 Sc5d−/− tissues at 1 and 21 days of age. We then bred the mice to obtain compound mutant mice. Lbr Sc5d−/−:Dhcr14 Sc5d−/− and Lbr Sc5d−/−:Dhcr14+/− died in utero. Lbr+/−:Dhcr14 Sc5d−/− mice appeared normal at birth but, by 10 days of age, were growth-retarded and neurologically abnormal (with ataxia and tremors) and, consistent with a demyelinating process, evidenced vacuolation and swelling of the myelin sheaths in the spinal cord upon pathology evaluation. We observed neither vacuolation nor swelling of the myelin sheaths in either Lbr Sc5d−/− or Dhcr14 Sc5d−/− mice. In contrast to Lbr Sc5d−/− mice, Lbr+/−:Dhcr14 Sc5d−/− mice had normal skin and did not display the Pelger-Huët anomaly. Peripheral tissue sterols were normal in all three mutant mice, although we found significantly elevated levels (50% of total sterols) of cholesta-8,14-dien-3β-ol and cholesta-8,14,24-trien-3β-ol in brain tissue from 10-day-old Lbr+/−:Dhcr14 Sc5d−/− mice. In contrast, we observed relatively small transient elevations in Δ14-sterol levels in Lbr Sc5d−/− and Dhcr14Δ4-7/Δ4-7 brain tissue. Our data support the idea that HEM dysplasia and ichthyosis result from impaired lamin B receptor function rather than from impaired sterol Δ14-reduction. Impaired sterol Δ14-reduction gives rise to a novel murine phenotype for which a corresponding human disorder has yet to be identified.
Niemann-Pick type C (NPC)
NPC is a neurodegenerative disorder that results in ataxia and dementia. In view of the dementia, it has been referred to as childhood Alzheimer's disease. The disorder is caused by a defect in intracellular lipid and cholesterol transport. Initially as part of a Bench-to-Bedside award, we began a clinical protocol to identify and characterize biomarkers that could be used in a subsequent therapeutic trial. The project also received support from the Ara Parseghian Medical Research Foundation and Dana's Angels Research Trust. We have enrolled over 55 NPC patients into a longitudinal Natural History trial. The goals of this trial are to identify (i) a blood-based diagnostic/screening test; (ii) biomarkers that can be used as tools to facilitate development and implementation of therapeutic trials; and (iii) clinical symptoms/signs that may be used as efficacy outcome measures in a therapeutic trial.
Currently, the average time from first symptom to diagnosis, the "diagnostic delay," in our cohort of NPC patients is on the order of four to five years. In collaboration with Dan Ory, we found elevated levels of nonenzymatically produced oxysterols in NPC patients. As well as being a potential a biomarker that may be used to follow therapeutic interventions, the oxysterols will likely form the basis of a blood-based diagnostic test that will significantly shorten the diagnostic delay encountered by NPC patients.
In addition to our Natural History study, we recently completed a randomized, placebo-controlled, cross-over trial to investigate the safety and efficacy of N-acetyl cysteine in NPC. The goal was to determine whether we could reduce oxidative stress and subsequently decrease levels of the non-enzymatically produced oxysterols. We are currently working with the Therapeutics of Rare and Neglected Disease Program of NHGRI to implement a therapeutic trial of cyclodextrin therapy in NPC.
To complement the clinical work, we have begun to apply molecular and proteomic approaches to both mouse and human biomaterials to identify biological pathways disrupted in NPC. We have identified several blood and cerebral spinal fluid proteins and we are in the process of validating the biomarkers as potential outcome measures to be used as tools in the development of therapeutic interventions.
Additional Funding
- NIH Director's Award (2009): Exome Sequencing in Autism Spectrum Disorder Patients with Altered Cholesterol Homeostasis
- Office of Rare Diseases and Ara Parseghian Medical Research Foundation (2009): Clinical Workshop on Neimann-Pick Disease, type C
- Ara Parseghian Medical Research Foundation (2009–2011): Niemann-Pick Disease, type C
- Autism Speaks (2009–2011): Double-masked, Placebo-controlled Trial of Cholesterol in Hypocholesterolemic ASD
- Bench to Bedside Program (2010–2011): Cyclodextrin Therapy for Niemann-Pick C1 Disease
- Bench to Bedside Program (2010–2011): Biomarkers of Neurodevelopment in Smith-Lemli-Opitz Syndrome
- Therapuetics for Rare and Neglected Diseases Program (2010–2012): Pilot Project on Niemann-Pick Disease, type C
Publications
- Yanjanin NM, Vélez JI, Gropman A, King K, Bianconi SE, Conley SK, Brewer CC, Solomon B, Pavan WJ, Arcos-Burgos M, Patterson MC, Porter FD. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet 2010;153B:132-140.
- Tierney E, Conley SK, Goodwin H, Porter FD. Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz Syndrome. Am J Med Genet A 2010;152A:91-95.
- Jiang X-S, Wassif CA, Backlund PS, Song L, Holtzclaw LA, Li Z, Yergey AL, Porter FD. Activation of Rho GTPases in Smith-Lemli-Opitz syndrome: pathophysiological and clinical implications. Hum Mol Genet 2010;19:1347-1357.
- Jiang X-S, Backlund PS, Wassif CA, Yergey AL, Porter FD. Quantitative proteomic analysis of inborn errors of cholesterol synthesis: identifcation of altered metabolic pathways in DHCR7 and SC5D deficiency. Mol Cell Proteomics 2010;9:1461-1475.
- Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS. Cholesterol oxidation producs are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med 2010;2:56ra81.
Collaborators
- Joan Bailey-Wilson, PhD, Inherited Diseases Research Branch, NHGRI, Bethesda, MD
- Yiannis Ioannou, PhD, Mount Sinai School of Medicine, New York, NY
- Norman Javitt, MD, PhD, New York University Medical School, New York, NY
- Daniel Ory, MD, Washington University, St. Louis, MO
- Marc Patterson, MD, Columbia University, New York, NY
- William Pavan, PhD, Genetic Disease Research Branch, NHGRI, Bethesda, MD
- Fran Platt, PhD, Oxford University, Oxford, UK
- Alan Remaley, MD, PhD, Molecular Diseases Branch, NHLBI, Bethesda, MD
- Gordon Watson, PhD, Children's Hospital Oakland Research Institute, Oakland, CA
- Alfred Yergey, PhD, Mass Spectrometry Core Facility, NICHD, Bethesda, MD
Contact
For more information, email fdporter@helix.nih.gov.