

You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Research Fellow
- Youn Hee Jee, MD, Senior Clinical Fellow
- Katerina Nella, MD, Clinical Fellow
- Ola Nilsson, MD, PhD, Clinical Fellow
We investigate the cellular and molecular mechanisms governing normal childhood growth and apply this knowledge to understand the causes of childhood growth disorders. In our basic science research, we focus on the mechanisms that allow rapid cell proliferation and hence rapid body growth in young children and that subsequently suppress proliferation, causing body growth to slow and eventually halt by adulthood. Elucidating the growth-limiting mechanisms not only provides insight into childhood growth disorders but may also have broader medical applications because disruption of these regulatory systems contributes to oncogenesis; conversely, their transient therapeutic suspension in adult cells might be used to achieve tissue regeneration. In our clinical research, we use high-throughput sequencing and other molecular-genetic approaches to study patients with growth disorders, including both growth failure and overgrowth.
Mechanisms by which estrogen limits childhood growth
Children grow taller because their bones get longer. The bone elongation occurs at the growth plate, a thin layer of cartilage within juvenile bones. We previously reported that the growth plate contains progenitor cells located within the resting zone. Children stop growing taller because the growth-plate cartilage undergoes programmed senescence, which involves extensive changes in gene expression, declining chonodrocyte proliferation, altered chonodrocyte differentiation, and involution of the growth plate. Eventually growth plate senescence leads to cessation of bone elongation and epiphyseal fusion. Estrogen accelerates this developmental process, causing growth to stop. We found that senescence occurs because progenitor cells in the resting zone of the growth plate are depleted in number and that estrogen acts by accelerating the depletion (2). The findings provide insight into the mechanisms that cause childhood linear growth to stop and into the endocrine regulation of this process; they also help define the mechanisms responsible for the early cessation of growth in children with precocious puberty, the late cessation of growth in individuals with hypogonadism, and the augmented adult height reported with aromatase inhibitor treatment of children.
Evolutionary conservation and modulation of a juvenile growth-regulating genetic program
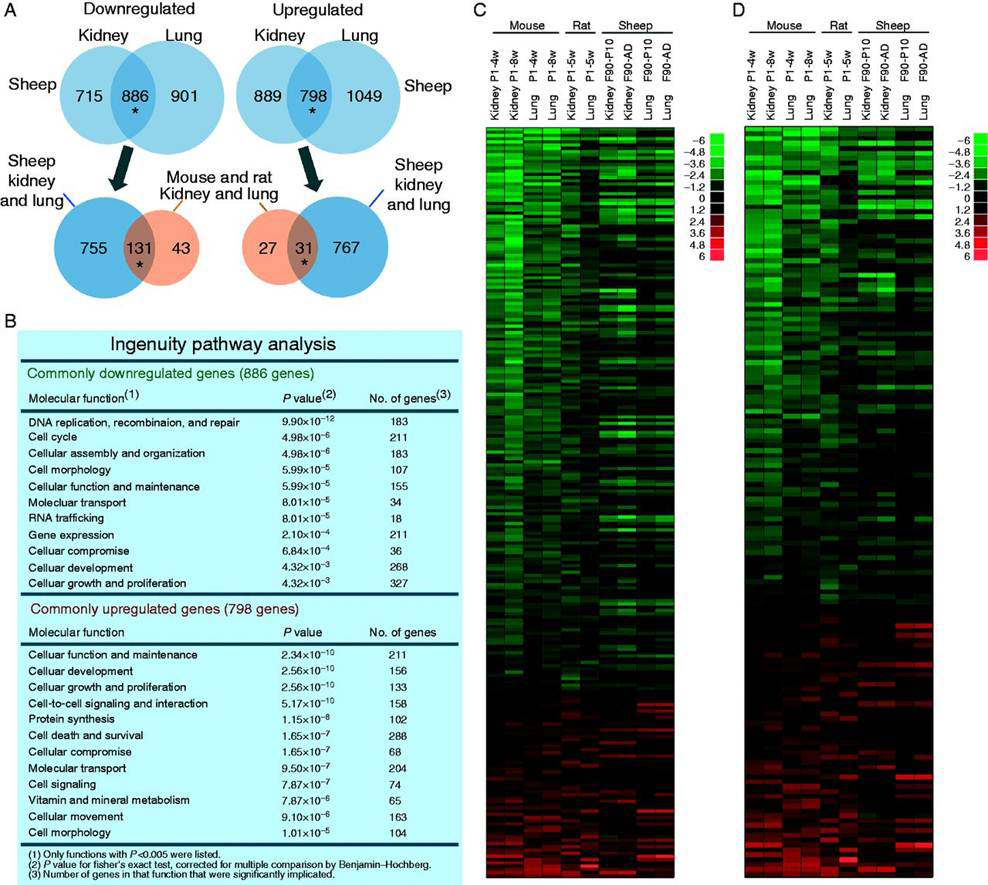
Click image to enlarge.
Figure 1. A multiorgan growth-regulating genetic program common to mice, rats, and sheep
A. Venn diagrams from microarray showing the number of genes significantly downregulated and upregulated with age in kidney and lung of the sheep vs. mouse and rat. *Overlap greater than expected by chance, P<0.001. B. Gene ontology analysis of the genes that were age-regulated in both organs in sheep. C. Heat map based on microarray analysis of the genes that were commonly down- or upregulated with age in the mouse and rat and present in the sheep microarray. Each row corresponds to a gene, ranked by average fold change in mouse and rat. D. Heat map similar to Panel C but excluding genes that have a known function specific to the cell cycle. In both heat maps, genes that were downregulated in the rodent tended to be downregulated in sheep and genes that were upregulated in rodent tended to be upregulated in sheep. Green, downregulation with age; red, upregulation. Scale values are log2 (fold difference). P, postnatal age (weeks); F, fetal age (days); AD, adult (21 months).
Body size varies enormously among mammalian species. In small mammals, body growth is typically suppressed rapidly, within weeks, whereas in large mammals, growth is suppressed slowly, over years, allowing for a greater adult size. We previously reported that body growth suppression in rodents is caused in part by a juvenile genetic program that occurs simultaneously in several tissues and involves the downregulation of a large set of growth-promoting genes. Recently, we found that the genetic program is conserved among mammalian species (Figure 1) but that its time course is evolutionarily modulated such that, in large mammals, it plays out more slowly, allowing for more prolonged growth and therefore greater adult body size (Figure 2) (3). The findings thus provide insight into the mechanisms underlying the enormous variation in body size among mammalian species. In addition, a deeper understanding of the mechanisms regulating body growth may have important clinical implications; in prior studies, we found that abnormalities of this program may contribute to disorders of childhood growth and to unrestricted growth in malignant cells.
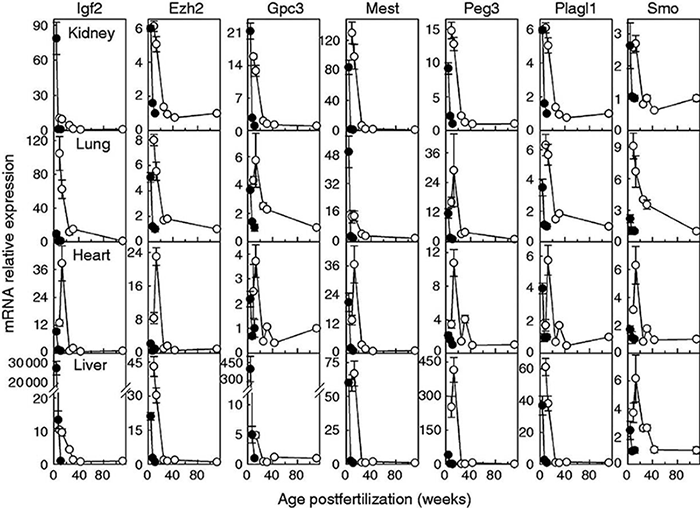
Click image to enlarge.
Figure 2. Downregulation of growth-regulating genes occurs more slowly in sheep (open circles) than in mice (closed circles).
Relative expression of mRNA (mean ± SEM) was assessed by quantitative RT-PCR. Top to bottom row: kidney, lung, heart, and liver. Relative expression was normalized to the young adult time point in each species (mouse, 11 weeks; sheep, 111 weeks).
Broad shifts in gene expression during early postnatal life are associated with shifts in histone methylation patterns.
We also explored epigenetic mechanisms that orchestrate the juvenile growth-regulating genetic program (4). Using chromatin immunoprecipitation–promoter tiling array, we found extensive genome-wide shifts in H3K4 and H3K27 histone trimethylation occurring with age in mice. We also assessed gene expression patterns by microarray analysis and found that H3K4 trimethylation is associated with high levels of gene expression and that H3K27 trimethylation is associated with low levels of gene expression in both kidney and lung of juvenile mice (Figure 3). Furthermore, changes in H3K4 trimethylation with postnatal age showed a strong, positive correlation with changes in gene expression, whereas changes in H3K27 trimethylation showed a negative association. Genes with decreases in H3K4 trimethylation with age were strongly implicated in cell-cycle and cell-proliferation functions. Taken together, the findings suggest that the common core developmental program of gene expression that occurs in multiple organs during juvenile life is associated with a common core developmental program of histone methylation. In particular, declining H3K4 trimethylation is strongly associated with gene downregulation and occurs in the promoter regions of many growth-regulating genes, suggesting that the change in histone methylation contributes to the component of the genetic program that drives juvenile body growth deceleration.
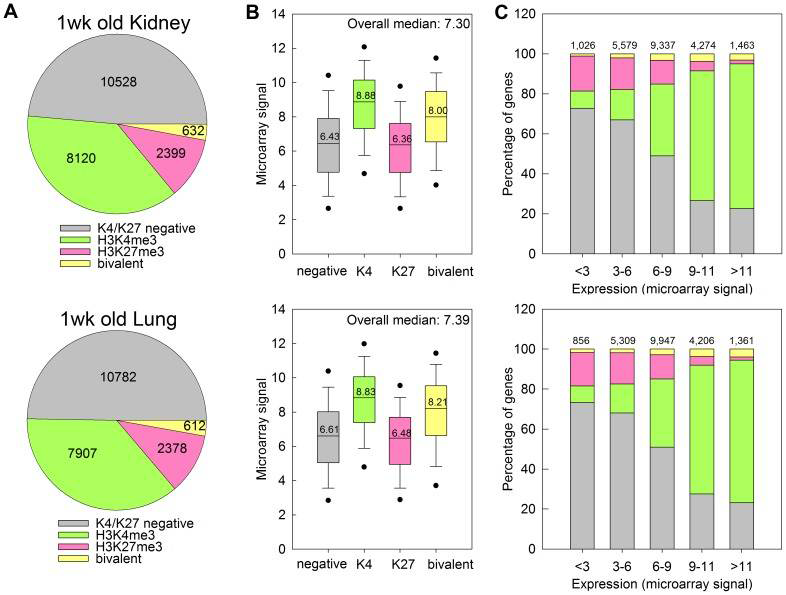
Click image to enlarge.
Figure 3. H3K4me3 is associated with high levels of gene expression and H3K27me3 is associated with low levels of gene expression in kidney and lung of 1-week-old mice.
A. Pie chart depicting the distribution of H3K4me3 and H3K27me3 marks at promoter regions of genes across the genome, in kidney (top) and lung (bottom) of 1-week-old mice. The numbers of genes within each category are indicated. B. Box and whisker plot showing the expression microarray signals of genes with different types of histone methylation. Genes with H3K4me3 showed higher expression, whereas genes with H3K37me3 showed lower expression levels. C. Bar graphs showing the distribution of H3K4me3 and H3K27me3 marks in gene sets with different levels of expression. H3K4me3 marks occurred more frequently in genes with higher expression levels, and H3K27me3 marks occurred more frequently in genes with lower expression levels. The color code for all three panels is shown in Panel A. Numbers at the top of bars represent total number of genes in each group.
Short stature with accelerated bone maturation due to aggrecan mutations
In some children with subnormal linear growth, a cause can be identified, but in many the etiology remains unknown. The condition, idiopathic short stature (ISS), can sometimes be severe. One goal of our group is to uncover the causes of this growth failure. Recently, we used whole-exome sequencing to study three families with autosomal dominant short stature, advanced bone age, and premature growth cessation (Figure 4). In the families, we identified novel heterozygous mutations in ACAN, which encodes aggrecan, a proteoglycan in the extracellular matrix of growth plate and other cartilaginous tissues (5). Our study demonstrated that heterozygous mutations in ACAN can cause a skeletal dysplasia that presents clinically as short stature with advanced bone age. The accelerating effect on skeletal maturation has not been previously noted in the few prior reports of human ACAN mutations. Our findings thus expand the spectrum of ACAN defects and provide a new molecular genetic etiology for the child who presents with short stature and accelerated skeletal maturation.
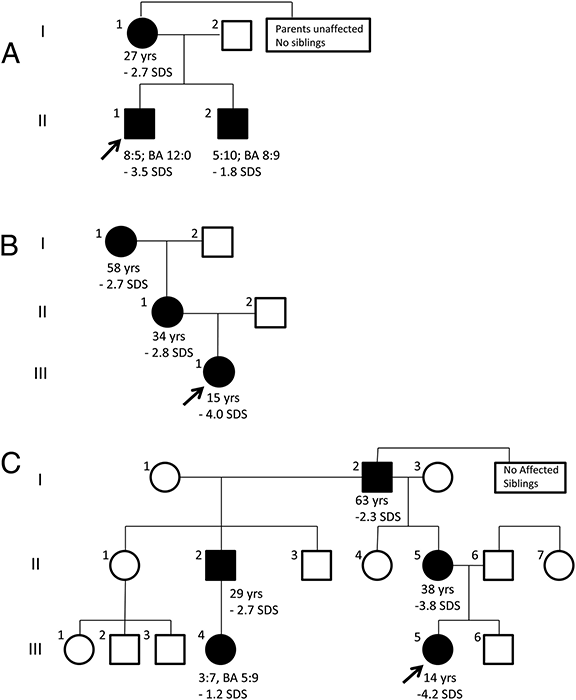
Click image to enlarge.
Figure 4. Pedigrees of families with autosomal dominant short stature and advanced skeletal maturation due to mutation in ACAN
Individuals carrying heterozygous ACAN mutations are indicated by solid symbols, whereas unaffected individuals are indicated as open symbols. The age (years) and adult height SDS (standard deviation score) is indicated for all affected individuals who have reached adult height and age (years:months), bone age (BA; years: month), and height SDS at most recent visit for all affected children who are still growing.
Additional Funding
- Pediatric Endocrine Society - Research Fellowship Award
- Thrasher Research Fund - Early Career Award
- Clinical Center Genomic Opportunity Program
Publications
- Lui JC, Baron J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc Natl Acad Sci USA 2013;110:6181-6186.
- Nilsson O, Weise M, Landman EB, Meyers JL, Barnes KM, Baron J. Evidence that estrogen hastens epiphyseal fusion and cessation of longitudinal bone growth by irreversibly depleting the number of resting zone progenitor cells in female rabbits. Endocrinology 2014;155:2892-2899.
- Delaney A, Padmanabhan V, Rezvani G, Chen W, Forcinito P, Cheung CS, Baron J, Lui JC. Evolutionary conservation and modulation of a juvenile growth-regulating genetic program. J Mol Endocrinol 2014;52:269-277.
- Lui JC, Chen W, Cheung CS, Baron J. Broad shifts in gene expression during early postnatal life are associated with shifts in histone methylation patterns. PLoS One 2014;9(1):e86957.
- Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J, Dauber A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab 2014;99:E1510-8.
Collaborators
- Andrew Dauber, MD, Boston Children’s Hospital, Boston, MA
- Dimiter Dimitrov, PhD, Laboratory of Experimental Immunology, Center for Cancer Research, NCI, Frederick, MD
- Joel Hirschhorn, MD, PhD, Harvard Medical School, Boston, MA
- Jan Maarten Wit, MD, Universiteit Leiden, Leiden, The Netherlands
- Ola Nilsson, MD, PhD, Karolinska Institute, Stockholm, Sweden
- Brigitte C. Widemann, MD, Pediatric Oncology Branch, Center for Cancer Research, NCI, Frederick, MD
- Jack Yanovski, MD, PhD, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.