

You are here: Home > Section on Developmental Genetics
Childhood Neurodegenerative Lysosomal Storage Disorders

- Anil Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Zhongjian (Gary) Zhang, MD, PhD, Staff Scientist
- Sondra W. Levin, MD, Adjunct Scientist
- Maria B. Bagh, PhD, Visiting Fellow
- Goutam Chandra, PhD, Visiting Fellow
- Vinay Patil, PhD, Postdoctoral IRTA fellow
- Shiyong Peng, PhD, Visiting Fellow
- Chinmoy Sarkar, PhD, Visiting Fellow
- Ashleigh Bouchelion, BS, MD/PhD Student
The Section on Developmental Genetics conducts both laboratory and clinical investigations into neurodegenerative lysosomal storage disorders predominantly affecting children. Our current laboratory research focuses on understanding the molecular mechanism(s) of pathogenesis of a group of hereditary childhood neurodegenerative lysosomal storage disorders (LSDs) called neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease. Mutations in at least 14 different genes underlie various types of NCLs, and the list of genes continues to grow. Currently, there is no effective treatment for any of the NCL types. The infantile NCL (or INCL) is an autosomal recessive disease caused by mutations in the CLN1 gene, which encodes palmitoyl-protein thioesterase-1 (PPT1), a lysosomal depalmitoylating enzyme. Numerous proteins undergo palmitoylation (S-acylation), a post-translational modification of polypeptides in which a fatty acid (predominantly palmitate) is attached to specific cysteine residues by thioester linkage. While the lipid modification regulates the function of many important proteins requiring membrane anchorage, the proteins must also be de-palmitoylated in order to recycle or undergo degradation by lysosomal hydrolases. Thus, PPT1 deficiency causes accumulation of palmitoylated proteins (constituents of ceroid) in the lysosomes, leading to INCL. However, the precise molecular mechanism of INCL pathogenesis remains largely unclear. Children afflicted with INCL are normal at birth, but exhibit signs of psychomotor retardation by 11 to 18 months of age. By two years of age, they are completely blind as a result of retinal degeneration and, by the age 4, manifest no brain activity and remain in a vegetative state for 6 to 8 years before eventual death. Such grim outcomes underscore the urgent need for the development of rational and effective therapeutic strategies not only for INCL but also for all NCLs. Thus, our goals are: (i) to understand the molecular mechanism(s) of pathogenesis through innovative laboratory investigations and (ii) to apply the knowledge gained from our studies to develop novel therapeutic strategies for INCL and possibly for other types of Batten diseases.
Studies in a Ppt1 knockout mouse model of INCL

Click image to enlarge.
Members of the Section on Developmental Genetics, PDEGEN.
Sitting from left to right: Ashleigh Bouchelion, Sondra W. Levin, Maria B. Bagh; standing from left to right: Shiyong (Sam) Peng, Goutam Chandra, Anil Mukherjee, Zhongjian (Gary) Zhang, and Vinay Patil
In our studies related to INCL, we use cultured cells from patients as well as Ppt1–knockout (Ppt1−/−) mice, which recapitulate virtually all clinical and pathological features of the disease. Previously, we found that PPT1 deficiency causes endoplasmic reticulum (ER) and oxidative stress, which at least in part contribute to neuropathology in INCL. Moreover, we delineated a mechanism by which PPT1 deficiency may disrupt the recycling of the synaptic vesicles (SVs), causing progressive loss of the SV pool size that is required for maintaining uninterrupted neurotransmission at nerve terminals. We also developed a noninvasive method, using MRI and MRS (magnetic resonance spectroscopy), to evaluate the progression of neurodegeneration in Ppt1−/− mice. The method allows repeated evaluations of potential therapeutic agents in treated animals. In addition, in collaboration with the NEI, we are conducting studies to determine whether electro-retinography can be used to assess the progressive retinal deterioration in Ppt1−/− as well as Ppt1–knock-in (KI) mice, which carry the most common nonsense mutation found in the U.S. INCL patient population. We also discovered that the blood-brain barrier is disrupted in Ppt1−/− mice and that this pathology is ameliorated by treatment with resveratrol, which has anti-oxidant properties.
Clinical trial using a combination of cysteamine bitartrate and N-acetylcysteine for INCL patients
Previously, we discovered that both cysteamine- and N-acetylcysteine–mediated ceroid depletion appeared to counteract the pathological changes in cultured cells from INCL patients. On the basis of these results, we conducted a bench-to-bedside clinical trial using a combination of the two drugs to determine whether the combination of oral cysteamine bitartrate and N-acetylcysteine is beneficial for patients with NCL. Children between 6 months and 3 years of age with INCL with any two of the seven most lethal PPT1 mutations were eligible for inclusion in this pilot study. All patients were recruited from physician referrals. Patients received oral cysteamine bitartrate (60 mg/kg per day) and N-acetylcysteine (60 mg/kg per day) and were assessed every 6-12 months until they had an isoelectric electroencephalogram (EEG, attesting to a vegetative state) or were too ill to travel. Patients were also assessed by electro-retinography, brain MRI and MRS, and electron-microscopic analyses of leukocytes for granular osmiophilic deposits (GRODs). Children also underwent physical and neurodevelopmental assessments on the Denver scale. Outcomes were compared with the reported natural history of INCL and that of affected older siblings. This trial was registered with ClinicalTrials.gov, number NCT00028262.
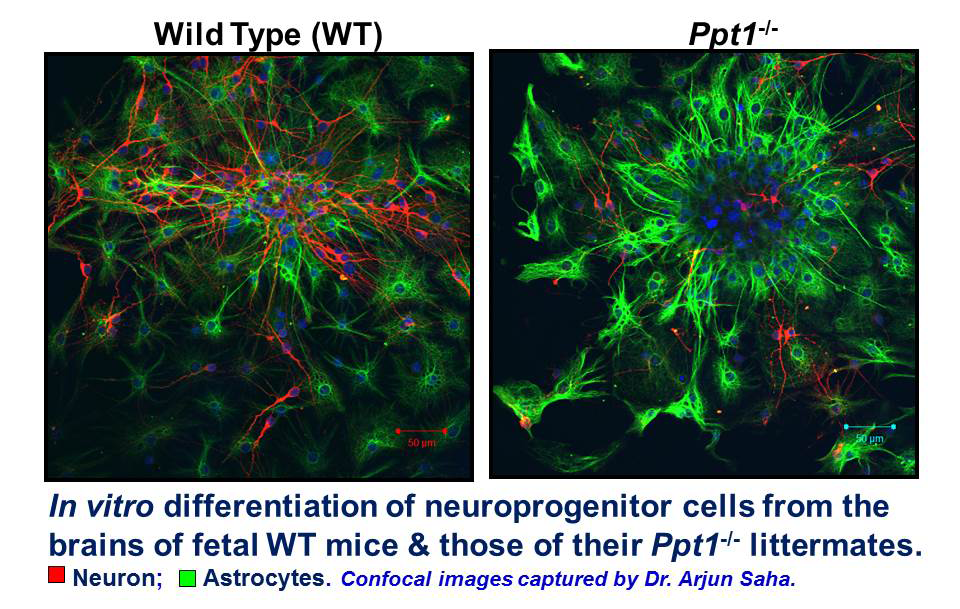
Click image to enlarge.
Figure 1. In vitro differentiation of neuroprogenitor cells from the brains of fetal WT mice and those of their Ppt1−/− littermates
Note that in Ppt1−/− neuroprogenitor cells in culture differentiate into more astrocytes than the ones from WT littermates, which differentiate into neurons and some astrocytes.
Between March 14, 2001, and June 30, 2012, we recruited ten children with INCL; one child was lost to follow-up after the first visit and nine patients (five girls and four boys) were followed for 8 to 75 months. MRI showed abnormalities similar to those in previous reports; brain volume and N-acetyl aspartic acid (NAA) decreased steadily, but no published quantitative MRI or MRS studies were available for comparison. None of the children acquired new developmental skills, and their retinal function declined progressively. Average time to isoelectric EEG (52 months, SD 13) was longer than reported previously (36 months). At the first follow-up visit, peripheral leukocytes in all nine patients showed virtually complete depletion of GRODs. Parents and physicians reported less irritability, improved alertness, or both in seven patients. No treatment-related adverse events occurred apart from mild gastrointestinal discomfort in two patients, which disappeared when liquid cysteamine bitartrate was replaced with capsules. The study was completed and the results recently published (1). Our findings suggest that combination therapy with cysteamine bitartrate and N-acetylcysteine is associated with delay of isoelectric EEG, depletion of GRODs, and subjective benefits, as reported by parents and physicians. Our systematic and quantitative report of the natural history of patients with INCL provides a guide for future assessment of experimental therapies.
A thioesterase-mimetic small molecule, N-tert-butyl hydroxylamine (NtBuHA), as a potential drug-target for INCL
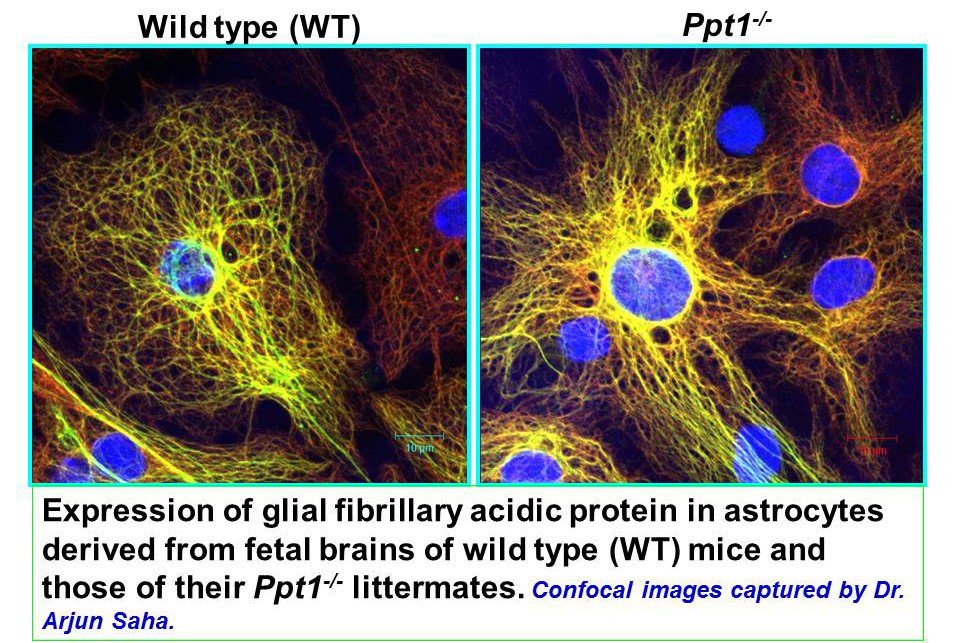
Click image to enlarge.
Figure 2. Astrocytes from wild type (WT) and Ppt1−/− mice
Localization of glial fibrillary acidic protein (GFAP) in astrocytes.
Palmitoylation (or S-acylation) has emerged as an important and the only reversible post-translational lipid modification of proteins. Thus, dynamic palmitoylation (palmitoylation-depalmitoylation) provides an important regulatory mechanism for the function of many proteins. Although palmitoylation is critical for protein function, depalmitoylation is equally important for these proteins to recycle or undergo degradation by lysosomal hydrolases. Although palmitoyl-acyl transferases (PATs), which catalyze palmitoylation of proteins, are being studied extensively, only a limited number of laboratories study the thioesterases. Because dynamic palmitoylation requires both PATs and thioesterases, we initiated studies on lysosomal PPT1 and acyl-protein thioesterase-1 (APT1), a cytosolic thioesterase. The studies aim to understand the potential cross-talk among the cytosolic and lysosomal thioesterases. Towards this goal, we had already made the unexpected discovery that the cytosolic thioesterases APT1 and APT2, which catalyze depalmitoylation of the proto-oncogene product of H-Ras and GAP-43, are themselves dynamically palmitoylated, promoting their cytosol membrane shuttling. Most interestingly, we found that APT1 catalyzes depalmitoylation of H-Ras as well as of APT2, while APT2 catalyzes depalmitoylation of GAP43. But most notably, we found that APT1 self-catalyzes its depalmitoylation, promoting its own dynamic palmitoylation possible (2).
Because the thioester linkage is labile and nucleophilic attack cleaves the linkage, we hypothesized that nucleophilic small molecules may have therapeutic potential for INCL. Hydroxylamine (HA), a potent nucleophilic metabolite present in all plants and animals, cleaves thioester linkage with high potency and specificity. Therefore, HA functionally mimics all thioesterases, including PPT1. However, at high concentrations, HA stimulates the production of methemoglobin, which, unlike hemoglobin, does not transport oxygen to the tissues, a toxicity that precludes its clinical application. We rationalized that derivatives of HA may be non-toxic while retaining the ability to cleave thioester linkage. We screened a panel of 12 HA derivatives and identified N-tert-butyl hydroxylamine (NtBuHA), which was non-toxic, cleaved thioester linkage in palmitoylated proteins, mediated depletion of lysosomal ceroid deposits and suppressed apoptosis in both cultured cells from INCL patients and in the brains of Ppt1−/− mice. Moreover, we found that NtBuHA was neuroprotective and modestly, albeit substantially, extended lifespan in the mice without elevating methemoglobin levels (3). Our findings provide a proof of principle that non-toxic, nucleophilic small molecules with antioxidant property, such as NtBuHA, are potential drug targets for diseases caused by thioesterase deficiency, such as INCL. U.S. and foreign patent applications for this invention are currently pending approval.
Mice homozygous for c.451C T mutation in the Cln1 gene recapitulate the INCL phenotype.
Nonsense mutations account for 5–70% of all genetic disorders. In the U.S., nonsense mutations in the CLN1/PPT1 gene underlie more than 40% of INCL cases. For evaluating nonsense suppressors in vivo, we sought to generate a reliable mouse model of INCL carrying the most common Ppt1 nonsense mutation (c.451C→T) found in the U.S. patient population. We knocked-in c.451C→T nonsense mutation in the Ppt1 gene in C57 embryonic stem (ES) cells using a targeting vector in which LoxP flanked the Neo cassette; the cassette was removed from targeted-ES cells by electroporating Cre. Two independently targeted ES clones were injected into blastocysts to generate syngenic C57 KI mice, obviating the necessity for extensive back-crossing. Generation of Ppt1-KI mice was confirmed by DNA sequencing, which showed the presence of c.451C→T mutation in the Ppt1 gene. The mice are viable and fertile, although they developed spasticity (a "clasping" phenotype) at a median age of six months. Autofluorescent storage materials accumulated throughout the brain regions and in visceral organs. Electron-microscopic analysis of the brain and the spleen showed granular osmiophilic deposits. Increased neuronal apoptosis was particularly evident in cerebral cortex, and abnormal histopathological and electro-retinographic (ERG) analyses attested to striking retinal degeneration. Progressive deterioration of motor coordination and behavioral parameters continued until eventual death. Our findings show that Ppt1-KI mice reliably recapitulate the INCL phenotype, providing a platform for testing the efficacy of existing and novel nonsense-suppressors alone or in combination with other drugs in vivo.
Publications
- Levin SW, Baker EH, Zein WM, Zhang Z, Quezado ZM, Miao N, Gropman A, Griffin KJ, Bianconi S, Chandra G, Khan OI, Caruso RC, Liu A, Mukherjee AB. Cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol 2014;13:777-787.
- Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, Mukherjee AB. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-Ras product and growth-associated protein-43. J Biol Chem 2013;288:9112-9125.
- Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1-/- mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci 2013;16:1608-1617.
- Bouchelion A, Zhang Z, Li Y, Qian H, Mukherjee AB. Mice homozygous for c.451C > T mutation in Cln1 gene recapitulate INCL phenotype. Ann Clin Transl Neurol 2014;1(12):1006-23.
Collaborators
- Eva Baker, MD, PhD, Radiology and Imaging Sciences, Clinical Center, NIH, Bethesda, MD
- Andrea Gropman, MD, Children's National Medical Center, Washington, DC
- Christopher J. McBain, PhD, Program in Developmental Neuroscience, NICHD, Bethesda, MD
- Kenneth Pelky, PhD, Program in Developmental Neuroscience, NICHD, Bethesda, MD
- Zenaide Quezado, MD, Children's National Medical Center, Washington, DC
- Ling-Gang Wu, PhD, Synaptic Transmission Section, NINDS, Bethesda, MD
Contact
For more information, email mukherja@exchange.nih.gov.