

You are here: Home > Section on Cellular Differentiation
Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Hyun-Sik Jun, PhD, Staff Scientist
- Chi-Jiunn Pan, BS, Senior Research Assistant
- Jun Ho Cho, PhD, Visiting Fellow
- Goo Young Kim, PhD, Visiting Fellow
- Joon Hyun Kwon, PhD, Visiting Fellow
- Young Mok Lee, PhD, Guest Researcher
- Brian C. Mansfield, PhD, Guest Researcher
We conduct research to delineate the pathophysiology of glycogen storage disease type I (GSD-I) and glucose-6-phosphatase-β (G6Pase-β or G6PC3) deficiency, and to develop novel therapies for these disorders. GSD-I consists of two subtypes, GSD-Ia, resulting from a deficiency in G6Pase-α (or G6PC) and GSD-Ib, from a deficiency in the glucose-6-phosphate (G6P) transporter (G6PT). A third disease, G6Pase-β deficiency, also known as severe congenital neutropenia syndrome type 4 (SCN4), is not a glycogen storage disease but biochemically a GSD-I–related syndrome (GSD-Irs). G6Pase-α and G6Pase-β are endoplasmic reticulum (ER)–bound G6P hydrolases, with active sites lying inside the lumen, which depend upon G6PT to translocate G6P from the cytoplasm into the ER lumen. The G6PT/G6Pase-α complex maintains interprandial glucose homeostasis while the G6PT/G6Pase-β complex maintains energy homeostasis and functionality of neutrophils and macrophages. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis not shared by GSD-Irs. GSD-Ib and G6Pase-β–deficient patients manifest a common myeloid phenotype of neutropenia and myeloid dysfunction not shared by GSD-Ia. Neutrophils express the G6PT/G6Pase-β complex, and inactivation of G6PT or G6Pase-β leads to the enhanced neutrophil apoptosis that underlies the neutropenia in GSD-Ib and GSD-Irs. We also showed that the G6PT/G6Pase-β complex is essential for energy homeostasis in neutrophils. A deficiency in either G6PT or G6Pase-β prevents recycling of ER glucose to the cytoplasm, leading to neutrophil dysfunction. There is no cure for GSD-Ia, GSD-Ib, or GSD-Irs. Animal models of the three disorders are available and are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy.
rAAV–mediated gene therapy prevents HCA and corrects metabolic abnormalities in GSD-Ia mice.
Because of the deficiency in G6Pase-α, GSD-Ia patients manifest impaired glucose homeostasis. While dietary therapies are sufficiently successful to enable patients to attain near normal growth and pubertal development, long-term complications, including hepatocellular adenoma (HCA) with malignant potential, persist in GSD-Ia patients. We examined the efficacy of liver G6Pase-α delivery mediated by rAAV8-GPE, a recombinant AAV pseudotype 2/8 vector expressing human G6Pase-α directed by the 2864-bp of human G6PC promoter/enhancer (GPE). A recent study showed that HCA develops in 100% of liver-specific G6pc-null mice 78 weeks after gene deletion. We therefore examined the disease risk for hepatic neoplasia in a long-term dose-ranging study using G6pc−/− mice. We showed that rAAV8-GPE–mediated gene transfer, deliberately titrated down to determine the minimum therapeutic dose, restoring 3–128% of normal hepatic G6Pase-α activity and producing 61–90% of normal endogenous glucose in G6pc−/−, was sufficient to maintain glucose homeostasis. The treated mice displayed normal hepatic fat storage, normal blood metabolite and glucose tolerance profiles, reduced fasting blood insulin levels, no evidence of hepatic abnormalities or HCA. Fasting hypoglycemia is the hallmark of GSD-Ia. It is promising that the rAAV8-GPE–treated mice were able to sustain a 24-hour fast, which is a stress test of the ability of the liver to maintain blood normoglycemia through glycogenolysis and gluconeogenesis catalyzed by the G6Pase-α/G6PT complex in the absence of dietary glucose. This correlated with an increase in hepatic G6PT mRNA expression and a corresponding increase in microsomal G6P uptake activity, leading to the production of low but sufficient glucose to maintain interprandial glucose homeostasis. Our results suggest that G6Pase-α gene transfer may offer a therapeutic approach to the management of human GSD-Ia.
The most efficacious gene therapy vectors for GSD-Ia reported to date are rAAV8-GPE and rAAV8-miGPE; the latter is a rAAV8 vector expressing human G6Pase-α directed by a shorter 382-bp minimal (m) GPE. We undertook a direct comparison between the efficacies of the rAAV8-GPE and the rAAV8-miGPE vectors. We showed that the rAAV8-GPE vector directs significantly higher levels of hepatic G6Pase-α expression, achieves greater reduction in hepatic glycogen accumulation, and leads to a better tolerance of fasting than does the rAAV8-miGPE vector, suggesting that the rAAV8-GPE vector is the better vector to take forward into clinical trials.
GSD-1a mice receiving gene therapy are protected against age-related insulin resistance and obesity.
rAAV-GPE−mediated gene therapy normalizes blood glucose homeostasis in G6pc−/− mice for up to 90 weeks. Those treated G6pc−/− mice that were titrated to express 3−63% of normal hepatic G6Pase-α activity (AAV-L/M mice) produced endogenous hepatic glucose levels that were 61−68% of those in control littermates, had a leaner phenotype, and exhibited fasting blood insulin levels more typical of young adult mice. We showed that the mice are protected against age-related obesity and insulin resistance, compared with control littermates. Pathway investigation shows that hepatic carbohydrate response element binding protein signaling, which improves glucose tolerance and insulin signaling, is activated in the AAV-L/M mice. Moreover, several longevity factors in the calorie restriction pathway, including the NADH shuttle systems, NAD+ concentrations, and the AMP–activated protein kinase/sirtuin 1/peroxisome proliferator–activated receptor-γ coactivator 1α pathway, are up-regulated in the AAV-L/M mice. Our results suggest that full restoration of wild-type G6Pase-α activity will not be required to confer benefit in clinical gene therapy trials for GSD-Ia. Moreover, moderate reduction of hepatic G6Pase-α activity can promote a leaner phenotype and prevent the development of an age-related decline in insulin sensitivity. Thus, G6Pase-α activity is an attractive pharmaceutical target for diseases with insulin resistance and obesity.
The SLC37 family of phosphate-linked sugar phosphate exchangers
The SLC37 family comprises four sugar-phosphate exchangers, SLC37A1, SLC37A2, SLC37A3, and SLC37A4, which are anchored in the ER membrane. The best-characterized family member is SLC37A4, better known as the G6PT. A deficiency in G6PT causes GSD-Ib. SLC37A1, SLC37A2, and G6PT function as phosphate (Pi)-linked G6P antiporters catalyzing G6P:Pi and Pi:Pi exchanges. The activity of SLC37A3 is unknown. The primary in vivo function of the G6PT protein is to translocate G6P from the cytoplasm into the ER lumen where it couples with either liver/kidney/intestine–restricted G6Pase-α or ubiquitously expressed G6Pase-β to hydrolyze G6P to glucose and Pi. As stated in the Introduction, the G6PT/G6Pase-α complex maintains interprandial glucose homeostasis, and the G6PT/G6Pase-β complex maintains neutrophil energy homeostasis and functionality. G6PT is highly selective for G6P and is competitively inhibited by cholorogenic acid and its derivatives. Neither SLC37A1 nor SLC37A2 can couple functionally with G6Pase-α or G6Pase-β, and the antiporter activities of SLC37A1 or SLC37A2 are not inhibited by cholorogenic acid. There are no known disease associations for SLC37A1, SLC37A2, or SLC37A3. Given that only G6PT matches the characteristics of the physiological ER G6P transporter involved in blood glucose homeostasis and neutrophil energy metabolism, the biological roles for the other SLC37 proteins remain to be determined.
Molecular mechanisms of neutrophil dysfunction in GSD-Ib
In addition to the enhanced neutrophil apoptosis that is the underlying cause of GSD-Ib neutropenia, patients also manifest neutrophil dysfunction of unknown etiology. Previously, we showed that G6PT interacts with the enzyme G6Pase-β to regulate the availability of G6P/glucose in neutrophils. Deficiency in G6Pase-β activity in neutrophils impairs both their energy homeostasis and functionality. We showed that G6PT–deficient neutrophils from human GSD-Ib patients are similarly impaired, an impairment that is characterized by decreased glucose uptake and reduced levels of intracellular G6P, lactate, ATP, and NADPH, while functional impairment is reflected in reduced neutrophil respiratory burst, chemotaxis, and calcium mobilization. We further showed that the expression and membrane translocation of the NADPH oxidase subunit p47phox are downregulated in G6PT−deficient neutrophils, explaining why respiratory burst activity is impaired. We also showed that the hypoxia-inducible factor-1α (HIF-1α)/peroxisome proliferators−activated receptor-γ (PPAR-γ) pathway, which directly affects neutrophil respiratory burst, chemotaxis, and calcium mobilization, is activated in G6PT−deficient neutrophils. Taken together, our results demonstrate that the underlying cause of neutrophil dysfunction in GSD-Ib is impaired neutrophil energy homeostasis and activation of the HIF-1α/PPAR-γ pathway. Insights into the etiology of neutrophil dysfunction in GSD-Ib should facilitate the development of novel therapies for this disorder.
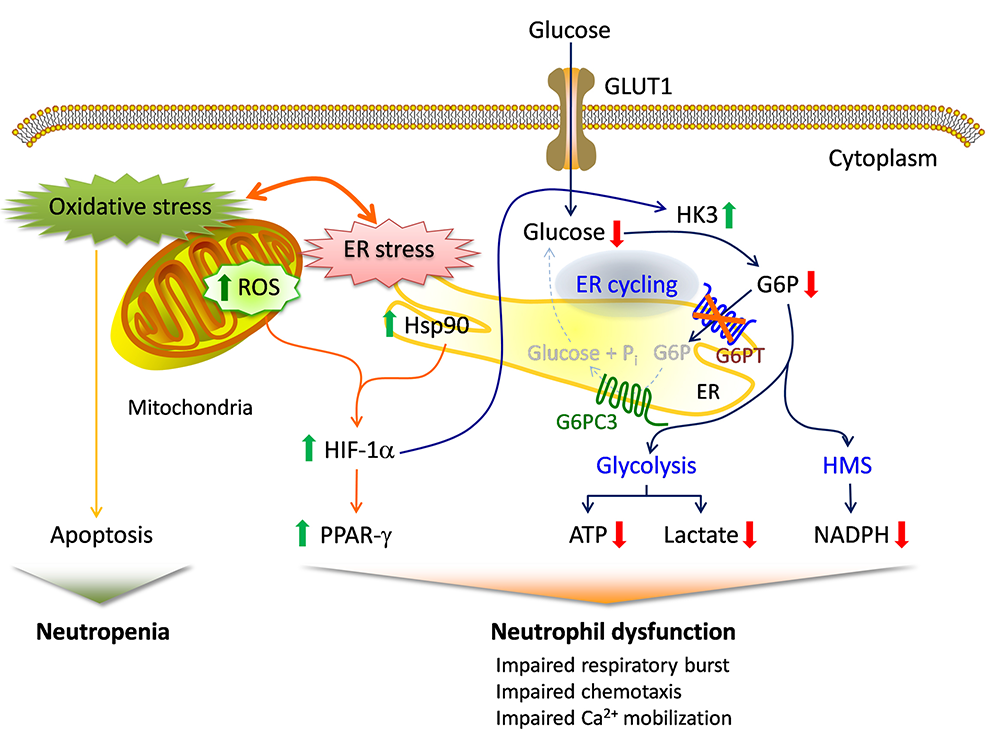
Click image to enlarge.
Proposed mechanisms underlying neutrophil dysfunction in GSD-Ib
Glucose transported into the cytoplasm via GLUT1 is metabolized by hexokinase (HK) to G6P, which participates in three major pathways: glycolysis, the hexose monophosphate shunt (HMS), and endoplasmic reticulum (ER) cycling. In cycling, G6P enters the ER via G6PT, where it can accumulate until it is hydrolyzed to glucose by G6Pase-β and transported back into the cytoplasm. By limiting cytoplasmic glucose/G6P availability, cycling regulates the other two cytoplasmic pathways for G6P metabolism. Disruption of ER cycling in G6PT–deficient neutrophils results in reduced glucose uptake and impaired energy homeostasis and thus functionality. The underlying cause of neutropenia in GSD-Ib is enhanced neutrophil ER stress and oxidative stress. The increases in Hsp90 and reactive oxygen species (ROS) in G6PT–deficient neutrophils stabilize HIF-1α (hypoxia-inducible factor-1α), an upstream activator of PPAR-γ (peroxisome proliferators−activated receptor-γ). The increase in PPAR-γ downregulates neutrophil respiratory burst, chemotaxis, and calcium mobilization activities. Thick arrows indicate the changes caused by a defect in G6PT activity.
Functional analysis of mutations in GSD-Irs
The G6Pase-β enzyme is embedded in the ER membrane and catalyzes the hydrolysis of G6P to glucose and phosphate. To date, 33 distinct G6Pase-β mutations have been identified in GSD-Irs patients but only the p.R253H and p.G260R missense mutations have been characterized functionally for pathogenicity. To demonstrate pathogenicity, we functionally characterized 16 of the 19 known missense mutations, using a sensitive assay based on a recombinant adenoviral vector-mediated expression system. Twelve missense mutations completely abolish G6Pase-β enzymatic activity while the p.M116V, p.T118R, p.S139I, and p.R189Q mutations retain, respectively, 1.1%, 1.3%, 49%, and 45% of wild-type G6Pase-β activity. A database of residual enzymatic activity retained by the G6Pase-β mutations will serve as a reference for evaluating genotype-phenotype relationships.
Additional Funding
- The Children's Fund for Glycogen Storage Disease Research, 2013
Publications
- Lee YM, Jun HS, Pan CJ, Lin SR, Wilson LH, Mansfield BC, Chou JY. Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology 2012;56:1719-1729.
- Chou JY, Jun HS, Mansfield BC. The SLC37 family of phosphate-linked sugar phosphate antiporters. Mol Aspects Med 2012;34:601-611.
- Lee YM, Pan CJ, Koeberl DD, Mansfield BC, Chou JY. The upstream enhancer elements of the G6PC promoter are critical for optimal G6PC expression in murine glycogen storage disease type Ia. Mol Genet Metab 2013;110:275-280.
- Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014;123:2843-2853.
- Chou JY, Mansfield BC. The SLC37 family of sugar-phosphate/phosphate exchangers. Curr Top Membr 2014;73:357-382.
Collaborators
- Barry J. Byrne, MD, University of Florida College of Medicine, Gainesville, FL
- Thomas Conlon, PhD, University of Florida College of Medicine, Gainesville, FL
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genoa, Italy
- Luigi Varesio, PhD, Istituto Giannina Gaslini, Genoa, Italy
- David A. Weinstein, MD, University of Florida College of Medicine, Gainesville, FL
Contact
For further information, contact chouja@mail.nih.gov.