

You are here: Home > Section on Molecular Endocrinology
Receptors and Actions of Peptide Hormones and Regulatory Proteins in Endocrine Mechanisms

- Maria L. Dufau, MD, PhD, Head, Section on Molecular Endocrinology
- Chon-Hwa Tsai-Morris, PhD, Staff Scientist
- Raghuveer Kavarthapu, PhD, Postdoctoral Fellow
- Peng Zhao, PhD, Postdoctoral Fellow
- Ruifeng Yang, MSc, Predoctoral Fellow
We investigate the molecular basis of peptide hormone control of gonadal function, with particular emphasis on the structure and regulation of the genes encoding the luteinizing hormone receptor (LHR) and prolactin (PRL) receptor (PRLR). We also investigate the regulatory mechanism(s) involved in the progress of spermatogenesis and the control of Leydig cell (LC) function. Our studies focus on the regulation of human LHR transcription (nuclear orphan receptors, epigenetics, DNA methylation, second messengers, repressors, corepressors, and coactivators), as well as on the multiple-promoter control of hPRLR gene transcription. We are elucidating the functions of two inhibitory short forms of prolactin receptors and their impact on the long form of the receptor as well as their relevance to physiological regulation and breast cancer. We also investigate novel gonadotropin-regulated genes relevant to the progression of testicular gametogenesis, LC function, and other endocrine processes. We focus on the function and regulation of the gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), an essential post-transcriptional regulator of spermatogenesis that was discovered, cloned, and characterized in our laboratory. The various functions of GRTH/DDX25 provide a fertile ground for the development of a male contraceptive.
The luteinizing hormone receptor
The luteinizing hormone receptor (LHR) is expressed primarily in the gonads, where it mediates luteinizing hormone (LH) signals that regulate cyclic ovarian changes or testicular function. Transcription of the LRH gene is regulated by complex and diverse networks, in which coordination and interactions between regulatory effectors are essential for silencing/activation of LHR expression. The proximal Sp1 site of the promoter recruits histone (H) deacetylases (HDAC) and the Sin3A corepressor complex, which contributes to the silencing of LHR transcription. Site-specific acetylation/methylation-induced phosphatase release serves as an on switch for Sp1 phosphorylation at Ser641, which causes p107 repressor release from Sp1, recruitment of transcription factor TFIIB and RNA polymerase II (Pol II), and transcriptional activation. Maximal derepression of the gene is dependent on DNA demethylation of the promoter, H3/H4 acetylation, and HDAC/Sin3 A release. Positive Cofactor 4 (PC4) has an important role in the assembly of the preinitiation complex (PIC) in trichostatin A (TSA)–mediated LHR transcription (1). PC4 is recruited by Sp1 following TSA treatment and acts as its coactivator. However, PC4 does not participate in TSA release of phosphatases, Sp1 phosphorylation, or release of repressor/complexes. Although TFIIB recruitment is dependent on PC4, we ruled out TFIIB as its direct target and acetylation of PC4 in the activation process. However, we demonstrated TSA–induced acetylation of PC4–interacting proteins, identified as acetylated H3 by mass spectroscopy; H3's presence in the complex in association with chromatin at the promoter was demonstrated by ChIP/reChIP. The role of these interactions in chromatin structure and their participation in the assembly of the PIC and transcriptional activation are under investigation.
Gonadotropin-regulated testicular RNA helicase
Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) is a testis-specific member of the DEAD-box family of RNA helicases present in LCs and meiotic germ cells. It is a multi-functional protein essential for the completion of spermatogenesis. Males lacking GRTH are sterile owing to the absence of sperm as a result of the failure of round spermatids to elongate. In addition to its intrinsic RNA helicase activity, GRTH is a shuttling protein that exports specific mRNAs from the nucleus to cytoplasmic sites. Our studies demonstrated that participation of the GRTH export/transport of mRNAs is essential for the structural integrity of the chromatoid body (an organelle that stores and processes mRNAs and resembles P-bodies in somatic cells) and the mRNAs' transit/association to actively translating polyribosomes, where GRTH may regulate translational initiation of genes. We identified mRNAs that are associated with GRTH and regulated at polysomal sites of spermatocytes and round spermatids of the mouse testis. The reduction in mRNAs associated at polysomal sites in the differential studies (knockout compared with wild type), which is not detected at the total cellular level but evident in the cytoplasm with abolition of protein expression, reflects the importance of the transport function of GRTH to relevant sites and underscore its impact on protein synthesis. The multiple functions of GRTH to regulate post-transcriptional events, including processing, exporting and storage of RNA, have been viewed as essential for controlling the availability of specific transcripts such as Tp1/2 and Prm 1/2 for translation during the progression of spermatogenesis. These chromatin remodelers, essential for spermatid elongation and completion of spermatogenesis, whose RNAs associate with GRTH, failed to express in GRTH–null mice with impaired mRNA nuclear export. We initiated studies to determine the RNA–binding motif(s)/regions within that GRTH protein that are required for association with germ-cell transition protein 2 (Tp2) mRNA transcripts. Two of the four conserved RNA binding motifs of the DEAD-box family of RNA helicases (Ia and V) are essential for GRTH binding to Tp2 3′ UTR mRNA; further studies on the characterization of Tp2 3′ UTR domains that interact with GRTH are in progress.
GRTH is regulated by LH through androgen at the transcriptional level in LCs (directly) and germ cells (presumably indirectly) of the testis, where GRTH's expression is both cell- and stage-specific. The helicase displays a novel negative autocrine control of androgen production in LCs by preventing overstimulation of the LH–induced androgen pathway through enhanced degradation of the StAR protein (steroidogenic acute regulatory protein) and, in this manner, controls the degree of cholesterol transport to the mitochondria and its availability for steroidogenesis. Our studies revealed the mechanism by which androgen and/or the androgen receptor (AR) regulates the expression of the GRTH gene in the LC via a short-range chromosomal loop. Through its activation of GRTH transcription, androgen/AR signaling in LCs participates in an autocrine regulatory mechanism with a major impact on LC steroidogenic function.
Our development of transgenic mice model carrying a Grth 5′ flanking region–GFP reporter provides a unique in vivo system for differential elucidation of regulatory regions upstream in the Grth gene that direct the gene's expression in germ cells (pachytene spermatocytes and round spermatids) and downstream in LCs (1). We identified a putative cis-binding element for germ cell–specific transcription factors (GCNF/RTR), predominantly expressed in developing germ cells (GC) and required for expression in the post-meiotic phase of GC of the testis, on −5270/−5262 within the distal 5′ flanking region of the Grth gene. By contrast, the proximal region directs basal Grth expression and androgen-induced intracrine expression in LCs through a functional androgen-response element (ARE). In the transgenic animal model, the AR antagonist flutamide blocked GRTH–GFP expression in LCs and germ cells, demonstrating direct (intracrine regulation by androgen/AR in LCs) and indirect effects of androgen/AR in GC, which do not express AR, through paracrine regulation by androgen/AR in Sertoli cells. Upon treatment with flutamide, GCNF protein expression was significantly reduced in the testis of transgenic mice carrying 6.4kb/3.6kb (the distal region required for cell-specific expression of Grth in GC), indicating the presence of a regulatory network for androgen GCNF–upstream 5′ UTR sequence to direct Grth expression in GC. ChIP analysis further demonstrated the association of GCNF with the GRTH sequence spanning the GCNF binding site; this interaction was abolished in round spermatids. The studies provide evidence for indirect androgen action on GCNF regulation of GRTH–specific expression in GC and insights for elucidation of intrinsic mediator(s) by androgen-specific function in Sertoli cells, which would in turn impact GCNF–induced expression of GRTH in GC. The model permits us to elucidate the mechanism of androgen action in GC and thus facilitate the identification of androgen-regulated factors that control expression of a critical gene(s) required for GRTH expression in GC involved in the progress of spermatogenesis. This could lead to development of contraceptive strategies in Sertoli cells to block sperm formation without impacting other aspects of androgen function.
The prolactin receptor
The PRL receptor (PRLR) is a member of the lactogen/cytokine receptor family, which mediates the diverse cellular actions of PRL. PRL is a major factor in the proliferation and differentiation of breast epithelium and is essential for stimulation and maintenance of lactation. It has been also implicated in the development of breast cancer, tumor growth, and chemo-resistance. In the human, PRLR expression is controlled at the transcriptional level by multiple promoters (one generic, [PIII], and five human-specific [hPN1-hPN5]), which were defined and characterized in our laboratory. Each promoter directs transcription/expression of a specific non-coding exon 1 (E1-3, hEN1-hEN5), a common non-coding exon 2, and coding exons (E3-E11). Transcription of PRLR in breast cancer cells is directed by the preferentially utilized PIII, which lacks an estrogen-responsive element. It is enhanced by estradiol (E2)/ERα (estrogen receptor α) through complex formation with the transcription factors Sp1 and C/EBPβ, which associate with cognate elements at their DNA sites, inducing transcription factor TFIIB and Pol II recruitment. Our bioluminescence resonance energy transfer (BRET) studies revealed ERα constitutive homodimers. Complex formation of the ERα dimer (non DNA bound) with Sp1 and C/EBPβ bound to their cognate sites is required for basal (constitutive ERα homodimers) and E2–induced transcriptional activation/expression of the human PRLR gene. PRL, which is highly expressed in tumoral breast, causes cell proliferation by activating its cognate receptor. Exacerbation of PRL's actions in breast cancer resulting from increase receptor expression can explain resistance to estrogen inhibitors in breast cancer. Our studies reveal that stimulation of PRLR transcription, mRNA, and protein in the human hormone-dependent breast cancer cell lines MCF7 and T47D by PRL in the absence of E2 is abolished by mutation of a GAS site (DNA-recognition motif) at the promoter (non-coding exon 1), small interfering RNA (siRNA) against the transcription activator Stat5, or by an ER antagonist (ICI). This indicates the participation of the ERα in PRLR transcription via PRL/PRLR/Stat5. PRL/PRLR induces phosphorylation of ERα through the JAK2/PI3K/MAPK//ERK and the HER2–activated pathways (Figure 1). The increased recruitment of phospho-ERα to Sp1 and C/EBPβ bound at their promoter sites is essential for PRL–induced receptor transcription. The recruitment is prevented by blockade of PRL expression or ERα phosphorylation. Our studies provide evidence for direct local actions of PRL independent of E2 in the up-regulation of PRLR transcription/expression by an activation loop between Stat5 and the ERα/Sp1/C/EBPβ complex, with requisite participation of signaling mechanisms (2). Incorporation of therapeutic agents that inhibit the function of the hormone PRL or the PRLR receptor with those targeting the various signaling pathways, including those resulting from activation of HER2, could improve effectiveness in reversing therapy resistance in breast cancer. Moreover, a combination therapy targeting ER and PRLR directly can offer an additional avenue to eliminate constitutive activation of ER and of PRLR by endogenous prolactin. Prolactin's role in the up-regulation of PRLR maximizes the action of the endogenous hormone. Our studies offer a mechanistically rational basis that could explain persistent invasiveness (progression and metastasis) fueled by prolactin in certain states refractory to adjuvant therapies and further the basis for the treatment of refractory states in ERα+ breast cancer.
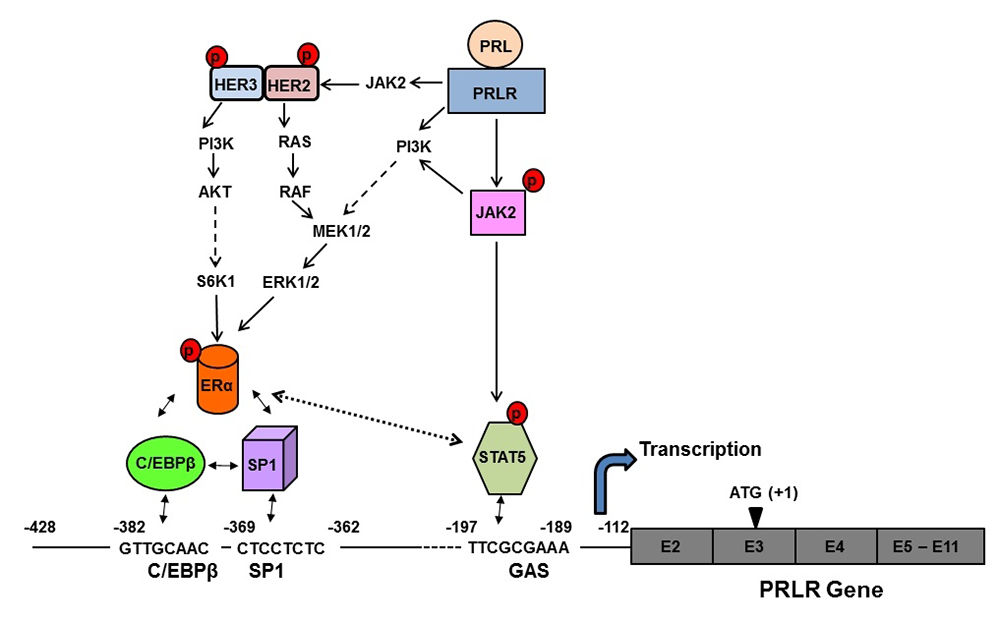
Click image to enlarge.
Figure 1. Mechanism of the up-regulation of the human prolactin receptor (hPRLR) induced by its cognate hormone in breast cancer cells
Prolactin (PRL) in normal and tumoral breast activates the JAK2/STAT5 pathway via the long form of the PRLR. The pSTAT5, bound to a GAS element located in the non-coding exon1 of the PRLR, associates with pERα (estrogen receptor construct) recruited to Sp1 and C/EBPβ (bound to their sites at the hPRLR hPIII promoter), causing increase hPRLR transcription and mRNA and protein production. PRL/PRLR, independent of estrogen, induces ERα phosphorylation (via the JAK2/PI3K-MEK-ERK and JAK2-HER2 pathways), nuclear translocation, complex formation by its recruitment to Sp1/C/EBPβ (bound to their DNA sites at the promoter), and transcriptional activation. The series of molecular events induced by endogenous PRL, by PRLR causing up-regulation of its cognate receptor, could participate in breast cancer progression and explain resistance to endocrine therapy.
Long-form (LF) homodimers of the PRLR mediate the diverse actions of PRL. The alternative spliced short form S1b (SF) inhibits the function of the LF through heterodimerization. A reduced S1b/LF-ratio in breast cancer could contribute to tumor development/progression. Our recent studies defined the structural and functional relevance of the D1 domain (most extracellular domain) of S1b to its inhibitory action on prolactin-induced LH function (3). The studies were conducted using mutagenesis, promoter/signaling analyses, BRET and molecular modeling. Mutation of E69 and adjacent residues at the receptor surface (S mutants) causes loss of its inhibitory effect, while mutations away from this region in D1 or in the D2 (domain most adjacent to membrane) (A mutants) displayed inhibitory action similar to that of the wild type. All mutants preserved PRL–induced activation of the tyrosine kinase JAK2. BRET revealed an increased affinity in D1–mutated S1b (S) homodimers favoring LF (homodimers)–mediated signaling induced by prolactin. By contrast, the affinity of the S1b homodimers in A and D2 remained unchanged (Figure 2). Molecular dynamics simulations show that mutations (S) elicit major conformational changes that propagate downward to the D1/D2 interface and change their relative orientation in the dimers. Major changes in receptor conformation and dimerization affinity are triggered by single mutations in critical regions of D1. The structure-function/simulation studies provide a basis for modeling and design of small molecules to enhance inhibition of LF activation for potential use in breast cancer and could also be applicable to other tumors, such as those of the prostate, in which endogenous PRL–mediated PRLR/Stat 5 activation has been correlated with high tumor grade and aggressive course of the disease, and in ovarian cancer, in which PRL–induced increases in survival and migration of ovarian cancer cells were effectively inhibited by S1b. Such compounds could also be used in cases of resistance to endocrine therapy, where in an autocrine fashion endogenous prolactin in the absence of estrogen could contribute to tumor growth and metastasis.
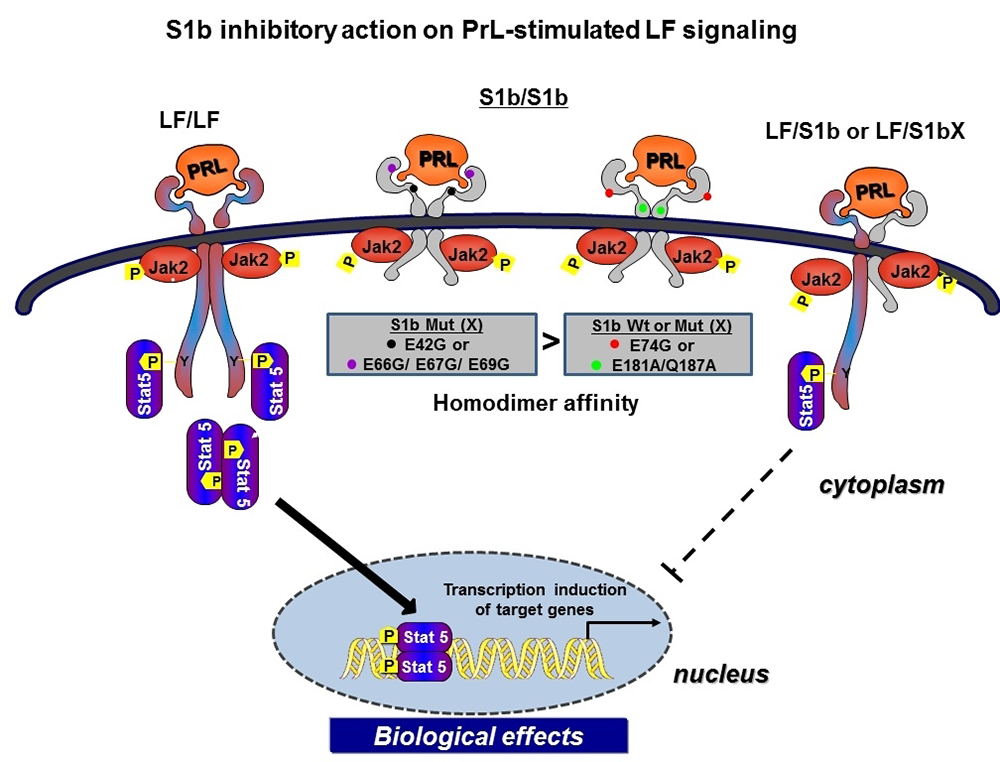
Click image to enlarge.
Figure 2. Mechanism of inhibitory action of wild-type and wild-type–like short-form PRL (S1b) on prolactin-induced long-form (LF) receptor signaling; non-inhibitory modality induced by key single mutations in S1b
Homodimers of the hPRLR form (LF) mediate the prolactin-stimulated JaK2/Stat5 signaling required for transcription/expression of PRL target genes, which are essential for the various biological effects of the hormone. The inhibitory action of short-form (SF) S1b on LF’s function results from LF/SF heterodimer formation and marked reduction of LF/LF homodimers, which are required by Stat 5 activation. PRL induces JaK2 phosphorylation (Jak2p) via LF/LF, LF/SF, and SF/SF dimers owing to the presence of Box1 (anchor of JaK2) in all forms. For signaling, i.e., the distal activation of Stat5 (Stat5-p), essential for transcriptional induction of PRL–responsive genes, to proceed, LF/LF dimers are required. Mutation (X) of amino acids located on the D1 domain adjacent to the hormone-binding domain (black and purple dots) reverses the inhibitory action of the wild-type S1b, while mutants located away from the hormone-binding pocket (red dot) and in the D2 domain (domain adjacent to the membrane, green dot) retain an inhibitory function similar to wild-type S1b. The loss of the inhibitory mode of S1bX results from differences in the affinity of homodimerization between the wild-type S1b and single amino acid mutants residing close to the binding pocket of S1b. The higher affinity observed in the S1b mutants in D1 close to the binding pocket and on the surface, compared with wild-type and like-wild type-mutants in D1 (away from the binding pocket and in D2), leads to LF/LF homodimerization with intact JaK2 signaling. The functional differences of S1b inhibitory action in wild type and mutants away from the pocket (inhibitory) and non-inhibitory action in mutants adjacent to the pocket were found, in modeling studies, to relate to conformational changes that propagate to the D1/D2 interface and to the relative change of the orientation of the dimers. Because the targeted amino acids are accessible to the solvent, hence to small molecules, they may offer critical sites of intervention for drug design and therapy in breast and other cancers.
Publications
- Kavarthapu R, Tsai Morris CH, Fukushima M, Pickel J, Dufau ML. A 5' flanking region of gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) gene directs its cell-specific androgen-regulated gene expression in testicular germ cells. Endocrinology 2013;154:2200-2207.
- Kavarthapu R, Tsai Morris CH, Dufau ML. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5. Oncotarget 2014;5:9079-9091.
- Kang JH, Hassan SA, Zhao P, Tsai Morris CH, Dufau ML. Impact of subdomain D1 of the short form S1b of the human prolactin receptor on its inhibitory action on the function of the long form of the receptor induced by prolactin. Biochim Biophys Acta 2014;1840:2272-2280.
Collaborators
- Sergio A. Hassan, PhD, Center for Molecular Modeling, Division of Computational Bioscience, NIH, Bethesda, MD
- James M. Pickel, PhD, Transgenic Core Facility, NIMH, Bethesda, MD
Contact
For more information, email dufau@helix.nih.gov.