

You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Postdoctoral Fellow
- Youn Hee Jee, MD, Clinical Fellow
- Katerina Nella, MD, Clinical Fellow
- Ola Nilsson, MD, PhD, Clinical Fellow
- Sao Fong Cheung, MPhil, Predoctoral Fellow
We investigate the cellular and molecular mechanisms governing childhood growth and development. We are especially interested in mechanisms that allow rapid cell proliferation and hence rapid body growth in young mammals and that subsequently suppress proliferation, thus setting a fundamental limit on the adult body size of the species. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. In addition, further investigation of the identified growth-limiting mechanisms may lead to broader medical applications, because disruption of these mechanisms may contribute to oncogenesis and, conversely, transient therapeutic suspension of growth-limiting mechanisms in adult cells might be used to achieve tissue regeneration.
Role of E2F transcription factors in the regulation of childhood growth
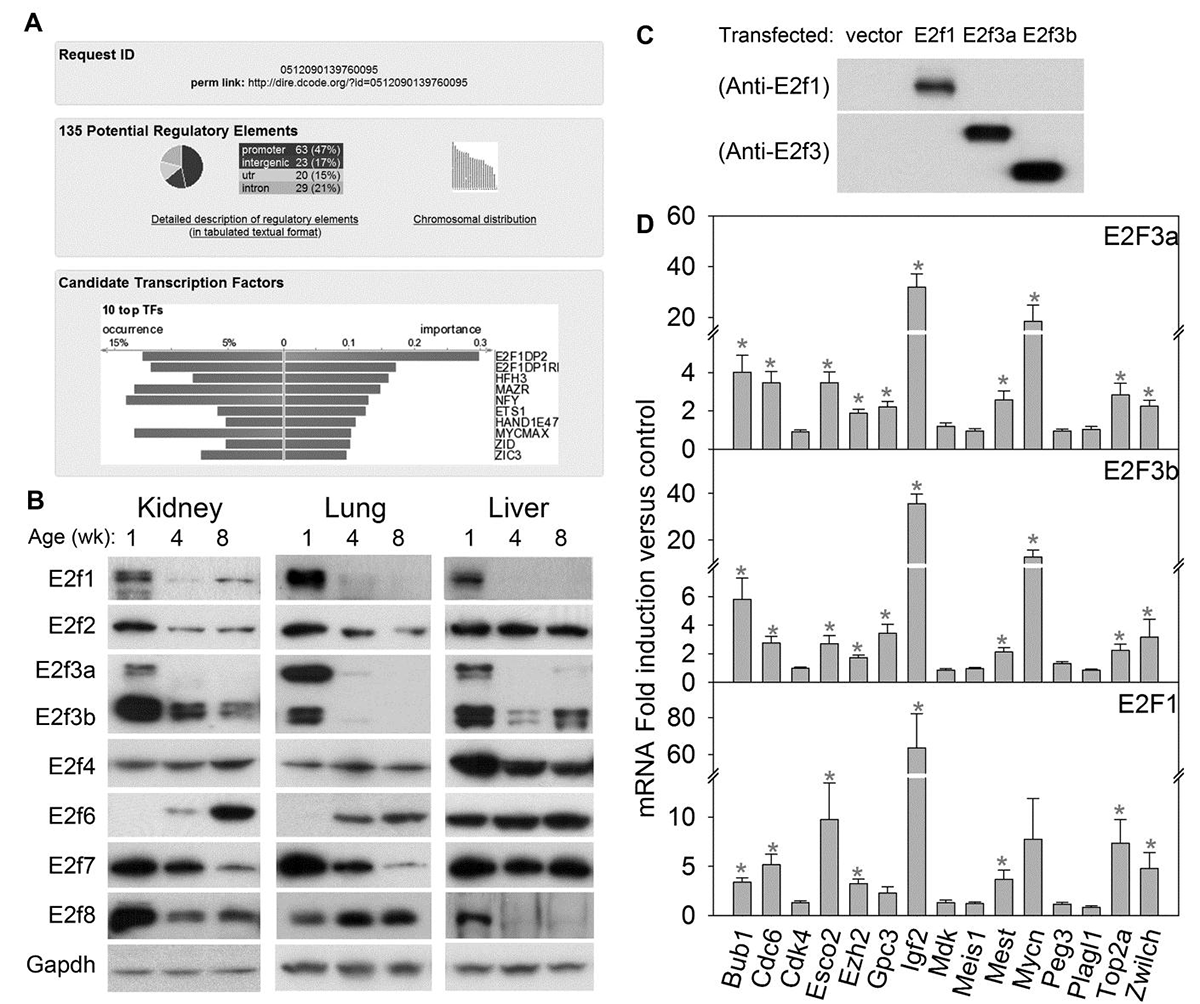
Click image to enlarge.
Figure 1. E2fs regulate many growth-promoting genes that are downregulated with age.
(A) Analysis of transcription factor binding sites using the bioinformatic program DiRE. The analysis compared a previously reported set of 235 genes that are downregulated with age in several organs during juvenile life with a random set of 5,000 genes to determine the most enriched binding sites. The bottom panel shows the top 10 candidate transcription factors implicated by the enriched regulatory elements, ranked by importance. (B) Western blot of E2f family of transcription factors in 1-, 4-, and 8-wk-old mouse kidney, lung, and liver. (C) Western blot of primary hepatocytes from 5-week-old mice transfected with empty vector or vectors expressing E2f1, E2f3a, or E2f3b. (D) Effect of E2fs on expression of 15 genes that are downregulated with age in several organs during juvenile life. Hepatocytes from 5-week-old mice were transfected with vectors expressing E2f1, E2f3a, or E2f3b. The effects on mRNA expression of the 15 genes were assessed by quantitative real-time PCR analysis. Expression levels were normalized to those of hepatocytes transfected with empty vector and shown as mean ± SEM. *, ANOVA P<0.05.
The human fetus grows at an enormous rate, increasing in mass more than 100-fold between the end of embryogenesis and birth. Somatic growth then slows progressively in postnatal life and ceases by the end of the second decade. The deceleration in body growth is primarily attributable to a progressive decline in cell proliferation, but the underlying mechanisms are largely unknown. We recently identified a juvenile multi-organ genetic program, which involves the downregulation of a large set of growth-promoting genes in mice and rats, and obtained evidence that the program helps explain the rapid body growth during early life and the subsequent slowing of growth with age.
We next sought to determine the molecular mechanisms that orchestrate the downregulation of this large gene set. Bioinformatic analysis indicated that E2f transcription factor–binding sites are strongly over-represented among genes participating in the growth-regulating genetic program (Figure 1A). We also found that transcription factors E2f1, E2f3a, and E2f3b are downregulated with age in several mouse organs (Figure 1B). Furthermore, restoration of high activator E2f expression in late juvenile murine hepatocytes helped restore high expression levels of several genes in the program (Figure 1C, D). Thus, the findings suggest that E2f transcription factors act as a molecular switch that helps orchestrate the juvenile growth-limiting program.
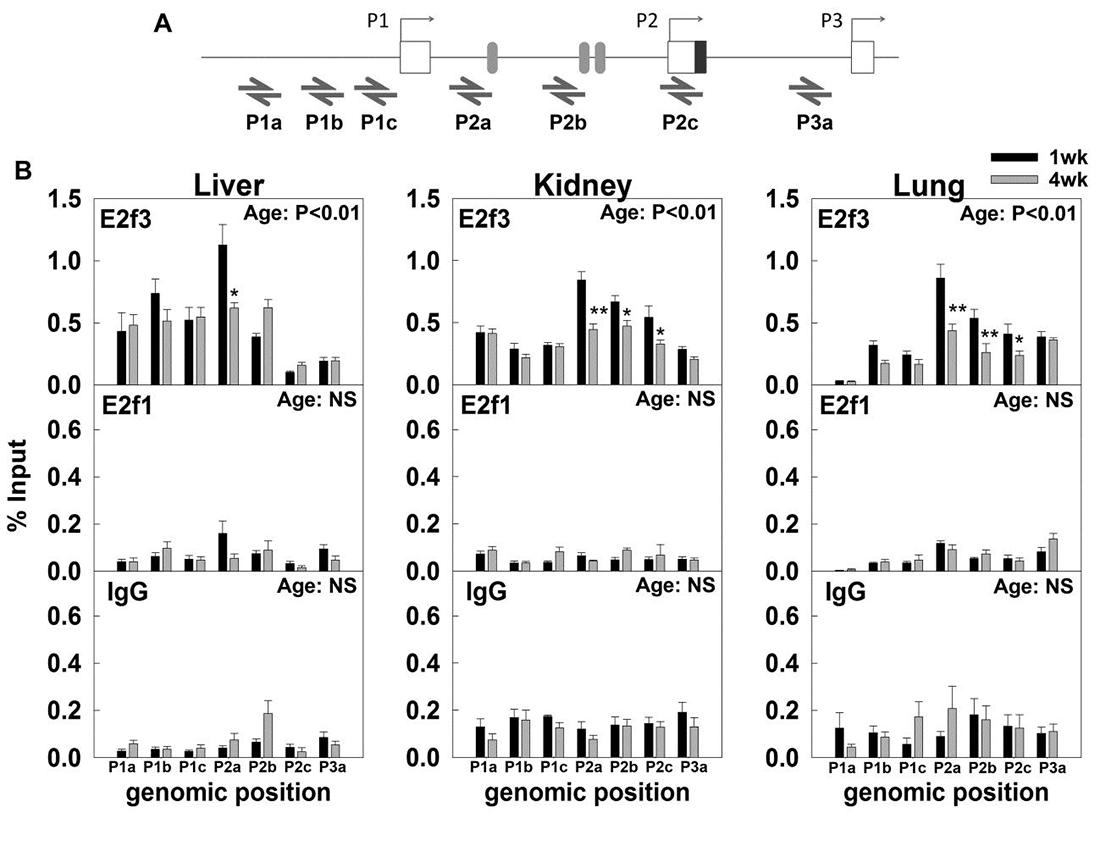
Click image to enlarge.
Figure 2. Changes in E2f3 and E2f1 binding to the Igf2 promoter from 1-week to 4-weeks of age in mouse liver, kidney, and lung.
We used chromatin immunoprecipitation (ChIP) assay to examine the binding of E2f3 and E2f1 to different Igf2 promoters. (A) Relative positions of primers used to quantify the E2f3 and E2f1 binding by real-time PCR. Boxes represent exons, and shaded ovals represent putative E2f binding sites predicted by Mulan/Multi-TF. (B) ChIP assay indicated that E2f3, but not E2f1, bound strongly to the mouse Igf2 P2 promoter but more weakly to the P1 and P3 promoter in 1-week-old organs. E2f3 binding to the P2 promoter declined from one week to four weeks of age in all three organs. IgG used as negative control. Two-way ANOVA was used to assess the effect of age and effect of genomic positions. P-value in top right corner of each graph, effect of age; data are shown as mean ± SEM; *, P<0.05, 1wk vs 4wk; NS, not significant.
We next focused on Igf2, an important gene in this program and which serves as a potent fetal growth factor. Mice with targeted Igf2 disruption exhibit severe growth retardation at birth. Similarly, in humans, reduced IGF2 expression is associated with poor fetal growth in the Silver-Russell syndrome, whereas elevated IGF2 expression is associated with excessive fetal growth and predisposition to tumors in the Beckwith-Wiedemann syndrome. Postnatally, IGF2 mRNA levels are downregulated in many organs. Thus, prior studies suggest that IGF2 is one of the growth-promoting genes that are highly expressed in early life, contributing to rapid body growth, but then are downregulated, causing growth to slow. We investigated the mechanisms responsible for the downregulation of IGF2 with age. Chromatin immunoprecipitation indicated that E2f3 specifically binds to the Igf2 P2 promoter region and that this binding declines with age (Figure 2). Overexpression of E2f3 strongly induced Igf2 expression (Figure 3) and could also activate reporter constructs containing the mouse Igf2 P2 promoter. Analysis of existing expression microarray data suggested that similar declines with age in E2F3 and IGF2 expression also occur in the human. Taken together, the findings suggest that downregulation of E2F3 with age contributes to the normal downregulation of IGF2, the gene encoding a key growth factor that controls the rate of body growth.
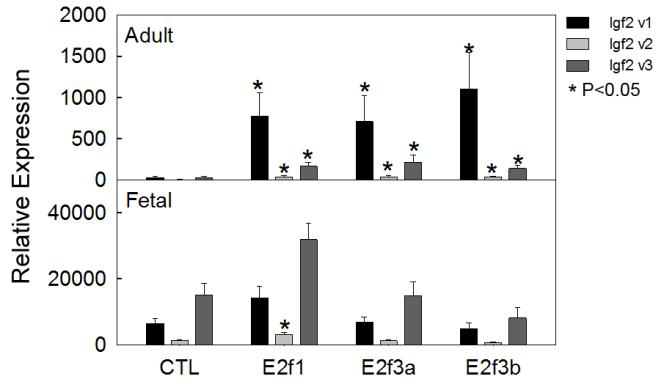
Figure 3. E2f3a and E2f3b induce Igf2 mRNA expression in late juvenile but not in fetal hepatocytes.
E2f3a and E2f3b expression vectors were transiently transfected into primary hepatocytes isolated from mice at five weeks of age (upper panel) or at embryonic day 15 (lower panel), and we measured Igf2 transcript variants two days later by real-time PCR. Variant 1 expression showed a more than 20-fold induction by E2f3a and E2f3b in adult hepatocytes, but not in fetal hepatocytes. Data are shown as mean ± SEM; v, variant; *, P<0.05 vs. control, ANOVA
Overexpression of IGF2 is commonly observed in childhood malignancies, as well as many cancers of adulthood. Thus, the high-level expression of IGF2 that occurs in fetal tissues and supports normal rapid body growth is often re-established in many malignant cells, perhaps contributing to their rapid proliferation. Based on our studies linking E2F and IGF2 expression in normal tissues, we hypothesized that the overexpression of IGF2 in some malignancies is a result of overexpression of E2Fs. Using microarray expression data, we found that, in E2F3–overexpressing prostate and bladder cancers, IGF2 expression is also elevated and positively correlated with E2F3 expression (Figure 4), providing evidence that the regulation of IGF2 by E2F3 may be an important mechanism for supporting rapid proliferation in cancer cells.
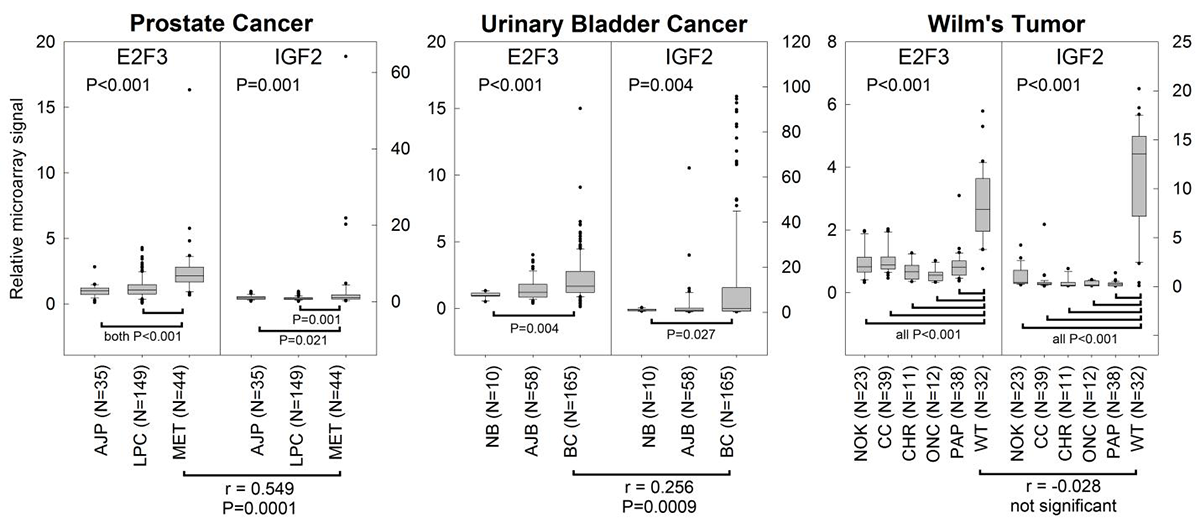
Click image to enlarge.
Figure 4. E2F3 overexpression correlates with IGF2 overexpression in prostate cancer and bladder cancer, but not Wilms' tumor.
Expression microarray data were gathered from the NCBI GEO database for three malignancies that overexpress E2F3: prostate cancer, bladder cancer, and Wilms' tumor. All three cancers showed greater expression of both E2F3 and IGF2 than did corresponding normal tissues. E2F3 and IGF2 levels were positively correlated in metastatic prostate cancers and bladder cancers (Spearman’s correlation, r-values and P-values near the bottom of graphs). AJP, normal prostate tissue adjacent to prostate cancer; LPC, local prostate cancer; MET, metastatic prostate cancer; NB, normal bladder mucosae; AJB, normal bladder mucosae adjacent to bladder cancer; BC, bladder cancer; NOK, normal kidney tissue; CC, clear cell renal cell carcinoma; CHR, chromophobe renal cell carcinoma; ONC, oncocytoma; PAP, papillary renal cell carcinoma; WT, Wilms' tumor. P-values (ANOVA) near top of graphs, overall effect of tissue type.
Identification of chondrocyte-binding peptides by phage display
There are currently no effective treatments for skeletal dysplasias. However, recent studies identified many paracrine factors that positively regulate growth plate chondrogenesis and therefore might be used to treat such disorders, including bone morphogenetic proteins, C-type natriuretic peptide, and Indian hedgehog. We reasoned that these locally acting molecules could be used therapeutically by administering them systemically but directing them to growth-plate cartilage using cartilage-targeting peptides. In addition, cartilage-targeting peptides could be used to direct endocrine therapy specifically to growth plate cartilage. For example, in achondroplasia, growth hormone increases bone length only modestly and dose is limited by effects on other tissues. Therefore, growth hormone linked to a cartilage-targeting peptide might augment the therapeutic growth-promoting effect and minimize adverse effects arising from actions on other tissues, including the potential for promoting malignancies.
As an initial step toward targeting cartilage tissue for potential therapeutic application, we sought cartilage-binding peptides using phage display, a powerful technology for selecting peptides that bind to molecules of interest. A library of phage displaying random 12–amino acid peptides was iteratively incubated with cultured chondrocytes to select phage that bind to cartilage. The resulting phage clones demonstrated enhanced affinity for chondrocytes by ELISA, when compared with a wild-type, "insertless" phage. Furthermore, the selected phage showed little preferential binding to other cell types, including primary skin fibroblast, myocyte, and hepatocyte cultures, suggesting a tissue-specific interaction. Immunohistochemical staining revealed that the selected phage bound to chondrocytes themselves and the surrounding extracellular matrix. We synthesized FITC–tagged peptides based on the sequence of cartilage-binding phage clones. These peptides, but not a random peptide, bound to cultured chondrocytes and extracelluar matrix. In conclusion, using phage display, we identified peptide sequences that specifically target chondrocytes. We anticipate that such peptides may be coupled to therapeutic molecules to provide targeted treatment for cartilage disorders.
Additional Funding
- Research Fellowship Award from the Pediatric Endocrine Society
Publications
- Lui JC, Nilsson O, Chan Y, Palmer CD, Andrade AC, Hirschhorn J, Baron J. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height. Hum Mol Genet 2012;21:5193-5201.
- Rezvani G, Lui JC, Barnes KM, Baron J. A set of imprinted genes required for normal body growth also promotes growth of rhabdomyosarcoma cells. Pediatr Res 2012;71:32-38.
- Dauber A, LaFranchi SH, Maliga Z, Lui C, Moon JE, McDeed C, Henke K, Zonana J, Kingman GA, Pers TH, Baron J, Rosenfeld RG, Hirschhorn JN, Harris MP, Hwa V. Novel microcephalic primordial dwarfism syndrome associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab 2012;97:E2140-2151.
- Lui JC, Baron J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc Natl Acad Sci USA 2013;110:6181-6186.
- Cheung CS, Lui JC, Baron J. Identification of chondrocyte-binding peptides by phage display. J Orthop Res 2013;31:1053-1058.
Collaborators
- Andrew Dauber, MD, Boston Children’s Hospital, Boston, MA
- Dimiter Dimitrov, PhD, Laboratory of Experimental Immunology, Center for Cancer Research, NCI, Frederick, MD
- Joel Hirschhorn, MD, PhD, Harvard Medical School, Boston, MA
- Brigitte C. Widemann, MD, Pediatric Oncology Branch, Center for Cancer Research, NCI, Frederick, MD
- Jan Maarten Wit, MD, Universiteit Leiden, Leiden, The Netherlands
- Jack Yanovski, MD, PhD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.