

You are here: Home > Section on Developmental Genetics
Hereditary Childhood Neurodegenerative Lysosomal Storage Disorders

- Anil Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Zhongjian (Gary) Zhang, MD, PhD, Staff Scientist
- Sondra W. Levin, MD, Adjunct Scientist
- Maria B. Bagh, PhD, Visiting Fellow
- Goutam Chandra, PhD, Visiting Fellow
- Vinay Patil, PhD, Visiting Fellow
- Shiyong Peng, PhD, Visiting Fellow
- Chinmoy Sarkar, PhD, Visiting Fellow
- Ashleigh Bouchelion, BS, MD/PhD Student
We conduct both laboratory and clinical investigations into hereditary neurodegenerative lysosomal storage disorders that mostly affect children. Our current laboratory research focuses on understanding the molecular mechanism(s) of pathogenesis of a group of hereditary childhood neurodegenerative lysosomal storage disorders (LSDs) called neuronal ceroid lipofuscinosis (NCL), also commonly known as Batten disease. Mutations in at least 13 genes cause various types of NCLs. Currently, there are no effective treatments for any of the NCL types. The infantile NCL (INCL) is an autosomal recessive disease caused by mutations in the CLN1 gene, which encodes palmitoyl-protein thioesterase-1 (PPT1), a lysosomal depalmitoylating enzyme. PPT1 deficiency causes accumulation of ceroid in lysosomes, leading to INCL pathogenesis. Children afflicted with INCL are normal at birth, but exhibit signs of psychomotor retardation by 11 to 18 months of age. By two years of age, they are completely blind due to retinal degeneration and, by the age 4, manifest no brain activity and remain in a vegetative state for 6 to 8 years before eventual death. These grim outcomes underscore the urgent need for the development of rational and effective therapeutic strategies not only for INCL but also for all NCLs. Thus, one of the major aims of this laboratory is to apply the knowledge gained from our investigations to develop novel therapeutic strategies for INCL and possibly for other Batten diseases.

Click image to enlarge.
Members of the Section on Developmental Genetics, PDEGEN.
Sitting from left to right: Ashleigh Bouchemion, Sondra W. Levin, Maria B. Bagh; standing from left to right: Shiyong (Sam) Peng, Goutam Chandra, Anil Mukherjee, Zhnogjian (Gary) Zhang, and Vinay Patil
Using Ppt1-knockout (Ppt1-KO) mice, which recapitulate virtually all clinical and pathological features of INCL, we discovered that PPT1 deficiency causes endoplasmic reticulum (ER) and oxidative stress, which at least contributes to neuronal death by apoptosis. We also delineated a mechanism by which PPT1 deficiency disrupts the recycling of the synaptic vesicles (SVs), causing progressive loss of SV pool size (2), which is required for maintaining uninterrupted neurotransmission at the nerve terminals. We also demonstrated that PPT1 deficiency disrupts adaptive energy metabolism and elevates ribosomal p-S6K1 levels, thus contributing to neuropathology in INCL (3). Moreover, we found that resveratrol, a naturally occurring anti-oxidant polyphenol, mitigates these abnormalities. During the past year, we developed a non-invasive method with which, by MRI and MRS, the progression of neurodegeneration in Ppt1-KO mice can be assessed, allowing repeated evaluations of potential therapeutic agents on treated animals. We also found that the blood-brain barrier is disrupted in Ppt1-KO mice and that this pathology is ameliorated by treating with resveratrol. We previously reported that both cysteamine and N-acetylcysteine counteract pathological changes in cultured cells from INCL patients. We concluded a bench-to-bedside study using a combination of these two drugs, and the results from this study are being analyzed. A major advance during the past year was the identification of a nucleophilic small molecule with antioxidant property that arrests neuropathology in Ppt1-KO mice and markedly extends their lifespan (4). A patent application has been filed for this invention.
During the past year, we made the unexpected discovery that the cytosolic thioesterases APT1 and APT2, which catalyze depalmitoylation of the protooncogene product of H-Ras and GAP-43 and promote their cytosol membrane shuttling, are palmitoylated at the N-terminal Cys-2. Moreover, we discovered that dynamic palmitoylation of APT1 and APT2 links that of their respective substrates. Further, we found that APT1 catalyzes depalmitoylation of H-Ras as well as of APT2, while APT2 catalyzes depalmitoylation of GAP43. Most importantly, we found that APT1 catalyzes its own depalmitoylation, making its own dynamic palmitoylation possible (1).
Our investigations continue to advance knowledge of INCL, paving the way for the application of the laboratory findings to the development of novel therapeutic approaches not only for INCL but also for other NCLs.
Dynamic palmitoylation links cytosol-membrane shuttling of APT-1 and APT-2 with that of H-Ras and GAP-43.
Palmitoylation (also called S-acylation) is the only reversible lipid modification in which a 16-carbon fatty acid (predominantly palmitate) is attached to cysteine residues of polypeptides via thioester linkage. Many soluble proteins require this modification for localization to membranes essential for function especially in the central nervous system. Although palmitoylation is required for membrane localization and function of these proteins, depalmitoylation is equally critical for recycling or for degradation by lysosomal hydrolases. Thus, dynamic palmitoylation (palmitoylation-depalmitoylation), like phosphorylation-dephosphorylation, is utilized as a cellular regulatory mechanism for protein function.
In mammals, palmitoylation is catalyzed by a family of 23 different enzymes called palmitoyl acyltransferases (PATs), whereas depalmitoylation is catalyzed by four thioesterases. Two of these thioesterases, acyl-protein thioesterase-1 (APT1) and APT2, are localized predominantly in the cytoplasm, and the other two, palmitoyl-protein thioesterase-1 (PPT1) and PPT2, are lysosomal enzymes. The first cytosolic thioesterase to be characterized was APT1, which catalyzes depalmitoylation of the proto-oncogene H-Ras product in vitro, as well as synaptosomal-associated protein 23 (SNAP-23) and the α-subunit of G-proteins. More recently, it was reported that a second cytosolic thioesterase, APT2, depalmitoylates GAP-43 (growth-associated protein 43). Although both H-Ras and GAP-43 undergo palmitoylation for membrane localization, these proteins also require depalmitoylation, catalyzed by APT1 and APT2 respectively, to detach from the membrane and to be recycled. However, until now it remained unclear how APT1 and APT2, being cytosolic enzymes, catalyze depalmitoylation of membrane-anchored, palmitoylated proteins such as H-Ras and GAP-43. The answer to this question is pivotal in understanding the roles of these proteins in health and disease. We hypothesized that both APT1 and APT2 undergo dynamic palmitoylation for steady-state membrane localization to catalyze depalmitoylation of their membrane-anchored, palmitoylated substrates, H-Ras and GAP-43, respectively.
We found that both APT1 and APT2 undergo palmitoylation on Cys-2. Moreover, blocking palmitoylation adversely affects membrane localization of both APT1 and APT2 and that of their substrates. We also demonstrate that APT1 not only catalyzes its own depalmitoylation but also that of APT2, promoting dynamic palmitoylation (palmitoylation-depalmitoylation) of both thioesterases. Furthermore, shRNA suppression of APT1 expression or inhibition of its thioesterase activity by palmostatin B markedly increased membrane localization of APT2; shRNA suppression of APT2 had virtually no effect on membrane localization of APT1. In addition, mutagenesis of the active-site Ser residue to Ala (S119A), which renders APT1 catalytically inactive, also increases its membrane localization. Taken together, our findings provide insight into a novel mechanism by which dynamic palmitoylation links cytosol–trafficking of APT1 and APT2 with that of their substrates, facilitating steady-state membrane localization and function of both.
Neuroprotection and lifespan extension in Ppt1-KO mice by NtBuHA: therapeutic implications for INCL
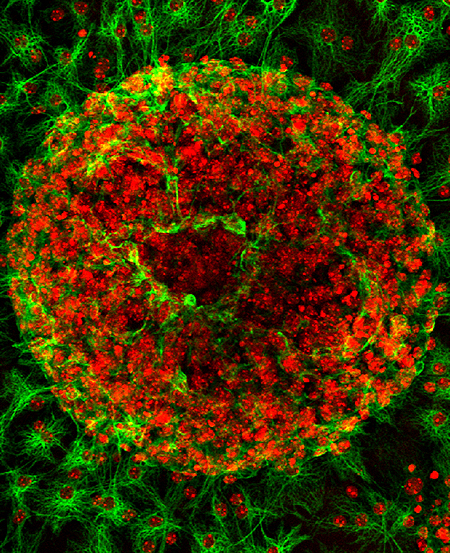
Click image to enlarge.
Figure 1.
In vitro differentiation of astrocytes (green) from neurospheres derived from a Ppt1−/− mouse fetal brain
Individually, lysosomal storage diseases (LSDs) are rare. However, cumulatively they affect 1 in 5,000–7,500 live births. Neurodegeneration is a devastating manifestation in the majority of the more than 50 LSDs. Neuronal ceroid-lipofuscinoses (NCLs), commonly known as Batten disease, constitute a group of the most common (1 in 12,500 births) genetically distinct, neurodegenerative storage disorders, which are characterized by progressive psychomotor deterioration with onset around early childhood to adolescence. Mutations in eight different genes underlie various types of NCLs, and several more NCL genes have been predicted to exist. Autofluorescent cytoplasmic inclusion granules in neurons and in other cell types are associated with selective destruction of neurons in the brain and in the retina. The NCLs as a group are broadly divided into two categories: one in which the gene mutations underlie a defect in a transmembrane protein and the other in which activities of soluble lysosomal enzymes are impaired. Notably, the deficiency of two soluble lysosomal enzymes, cathepsin D and PPT1, underlies two of the most lethal NCLs: congenital NCL and INCL, respectively.
The development of effective therapies remains challenging. As noted in the introduction, inactivating mutations in the CLN1 gene, which encodes the lysosomal depalmitoylating enzyme PPT1, cause lysosomal ceroid accumulation. PPT1 cleaves thioester linkage in palmitoylated (S-acylated) proteins (constituents of ceroid), facilitating degradation by lysosomal hydrolases. Because the thioester linkage is labile and is disrupted by nucleophilic attacks, we hypothesized that nucleophilic compounds may have therapeutic potential for INCL. Hydroxylamine (HA), a naturally occurring nucleophilic small molecule, cleaves thioester linkage with high specificity and functionally mimics PPT1. However, clinical application of HA is precluded because it stimulates production of methemoglobin, which unlike hemoglobin, does not transport oxygen to the tissues. While some HA derivatives are non-toxic, it is not known whether such compounds cleave thioester linkage in palmitoylated proteins and could therefore mediate ceroid depletion in INCL. Thus, we first screened a panel of HA derivatives for their ability to cleave thioester linkage in [14C]palmitoyl-CoA, a model thioester substrate of PPT1. We identified an HA derivative, N-tert-butyl HA (NtBuHA), which efficiently cleaves thioester linkage in both [14C]palmitoyl-CoA and, in palmitoylated proteins in cultured cells from INCL patients, mediates ceroid depletion, inhibits apoptosis, and is non-toxic. These beneficial effects in cellulo are completely reproducible in Ppt1-knockout mice. Moreover, NtBuHA treatment of these mice depletes ceroid, suppresses neuronal apoptosis, relieves neurological deterioration, does not stimulate methemoglobin production, crosses the blood-brain barrier, and markedly extends their lifespan. Our findings provide proof of principle that nucleophilic small molecules such as NtBuHA have therapeutic potential for INCL as well as for other as yet unrecognized thioesterase-deficiency diseases.
In a mouse model of Batten disease, palmitoyl protein thioesterase-1 deficiency is associated with brown adipose tissue and thermoregulation abnormalities.
We had previously shown that children with INCL have increased risk of hypothermia during anesthesia and that Ppt1 deficiency in mice is associated with disruption of adaptive energy metabolism, downregulation of peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α), and mitochondrial dysfunction. We hypothesized that Ppt1-knockout mice, the well-studied model of INCL that shows many of the neurologic manifestations of the disease, would recapitulate the thermoregulation impairment observed in children with INCL. We also hypothesized that, when exposed to cold, Ppt1-knockout mice would be unable to maintain body temperature, given that, in mice, thermogenesis requires upregulation of Pgc-1α and uncoupling protein 1 (Ucp-1) in brown adipose tissue. We found that the Ppt1-KO mice had lower basal body temperature as they aged and developed hypothermia during cold exposure. Surprisingly, this inability to maintain body temperature during cold exposure in Ppt1-KO mice was associated with an adequate upregulation of Pgc-1α and Ucp-1 but with lower levels of sympathetic neurotransmitters in brown adipose tissue. In addition, during baseline conditions, brown adipose tissue of Ppt1-KO mice had less vacuolization (lipid droplets) than did wild-type animals. After cold stress, lipid droplets in wild-type animals declined significantly whereas in Ppt1-KO the changes were insignificant compared with baseline measurements, suggesting that Ppt1-KO had less lipolysis in response to cold stress. The results uncover a previously unknown phenotype associated with PPT1 deficiency, that of altered thermoregulation, which is associated with impaired lipolysis and neurotransmitter release to brown adipose tissue during cold exposure. Our findings suggest that INCL should be added to the list of neurodegenerative diseases that are linked with alterations in peripheral metabolic processes and, furthermore, that impaired thermoregulation and hypothermia are potential risks in patients with INCL.
A bench-to-bedside study to determine whether a combination Cystagon™ and Mucomyst© is beneficial for patients with INCL
Given that PPT1 catalyzes the cleavage of thioester linkages in palmitoylated (S-acylated) proteins (see above), that its deficiency impairs degradation of these lipid-modified proteins by lysosomal proteases, that the consequent accumulation of these modified proteins (ceroids) in lysosomes leads to INCL pathogenesis, and the susceptibility of thioester linkages to nucleophilic attack, drugs that can cleave such linkages may have therapeutic potential for INCL. We showed that two drugs, phosphocysteamine and N-acetylcysteine, cleave thioester linkages in [14C] palmitoyl-CoA, a model substrate of PPT1, in lymphoblasts and fibroblasts from INCL patients. The drugs also facilitated the depletion of lysosomal ceroids and inhibited apoptosis. The results prompted us to initiate a bench-to-bedside clinical protocol to determine whether a combination of Cystagon TM (cysteamine bitartrate) and Mucomyst© (N-acetylcysteine) is beneficial for patients with INCL. Initially, the NICHD-IRB approved our protocol for the treatment of 20 INCL patients; the study has now been extended to include 40 INCL patients carrying any two mutations in the CNL1 gene. So far, we have treated 10 INCL patients; the study is currently recruiting patients and will continue until the approved number of patients is treated. The protocol admits INCL patients who are 6 months to 3 years of age. Patients may be referred to Dr. Mukherjee at mukherja@exchange.nih.gov.
Essential role(s) of PPT1 in lysosomal targeting and processing of pro-cathepsin D to mature cathepsin D
Lysosomes are membrane-bound organelles that maintain an acidic pH, required for activity of 50–60 acid hydrolases. Mutations in the genes encoding these proteins underlie various lysosomal storage diseases (LSDs). Cathepsin D (CD) (EC 3.4.23.5), a major lysosomal aspartyl protease, catalyzes the degradation and clearance of long-lived intracellular proteins. In the central nervous system, many of these proteins are palmitoylated. Failure to clear these long-lived intracellular proteins (many of which may be palmitoylated) causes accumulation, contributing to the pathogenesis of many neurodegenerative disorders. Paradoxically, it has been reported that both CD overexpression as well as CD deficiency underlie neurodegenerative diseases, although until now this paradox remained unresolved. We used Ppt1-KO mice to investigate this paradox. We found that in the brain of these mice CD is overexpressed. However in lysosomal fractions of the same tissues there is a relative CD deficiency. We also found that the endosomal/lysosomal targeting and maturation of pro-CD is disrupted as a result of impaired recycling of palmitoylated pro-CD receptors. Moreover, while pro-CD bound to palmitoylated receptors is transported to late endosome, elevated pH impaired receptor–ligand dissociation. Consequently, pro-CD bound to its receptors are transported to the lysosome where suppressed cathepsin B and L activities, required for processing pro-CD to mature-CD in lysosome, is impaired. Our findings provide insight into a paradox that despite CD overexpression, impaired endosomal/lysosomal targeting of pro-CD may cause lysosomal CD deficiency and neurodegeneration.
Additional Funding
- Batten Disease Support and Research Association (BDSRA)
Publications
- Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, Mukherjee AB. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-Ras product and growth-associated protein-43. J Biol Chem 2013;288:9112-9125.
- Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, Mukherjee AB. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest 2008;118:3075-3086.
- Wei H, Zhang Z, Saha A, Peng S, Chandra G, Quezado Z, Mukherjee AB. Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: partial rescue by resveratrol. Hum Mol Genet 2012;20:1111-1121.
- Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1−/− mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci 2013;in press.
Collaborators
- Eva Baker, MD, PhD, Warren Grant Magnuson Clinical Center, NIH, Bethesda, MD
- Andrea Gropman, MD, Children's National Medical Center, Washington, DC
- Zenaide Quezado, MD, Children's National Medical Center, Washington, DC
- Ling-Gang Wu, PhD, Synaptic Transmission Section, NINDS, Bethesda, MD
- Jianhua Xu, PhD, Georgia Health Sciences University, Augusta, GA
Contact
For more information, email mukherja@exchange.nih.gov.