



You are here: Home > Section on Eukaryotic DNA Replication and Gene Regulation
Regulation of Mammalian Cell Proliferation and Development

- Melvin L. DePamphilis, PhD, Head, Section on Eukaryotic Gene Regulation
- Christelle De Renty, PhD, Visiting Fellow
- Kotaro Kaneko, PhD, Staff Scientist
- Chrissie Lee, PhD, Postdoctoral Fellow
- Haiyan Pan, PhD, Postdoctoral Fellow
- Zakir Ullah, PhD, Visiting Fellow
- Alex Vassilev, PhD, Staff Scientist
- Xiaohong Zhang, BA, Technical Assistant
- Wenge Zhu, PhD, Postdoctoral Fellow
DNA replication origins are determined by genetic as well as epigenetic features. Genetic features are embedded in the DNA sequence itself and are generally protein binding sites or genes that encode the structure for specific proteins and RNAs. Epigenetic features are not determined by DNA sequence but are nevertheless heritable from one generation to the next. Moreover, epigenetic features are reversible and include chromatin structure, nuclear organization, transcription, transcription factors, deoxyribonucleotide pools, DNA topology, and DNA methylation. Consequently, the number and locations of replication origins in the cells of multicellular organisms can change from an average of one every 10 to 20 kb in the rapidly cleaving embryos of frogs, flies, and fish to one every 50 to 300 kb in the differentiated cells of adult organisms. Developmental changes in origin density also occur during specific stages in animal development. Thus, metazoan genomes contain many potential initiation sites for DNA replication, but, during development, some of these sites are selectively activated while others are suppressed. When we introduced the initiation/suppression concept many years ago, we termed it the “Jesuit Model” because the Jesuits remind us that while many are called, few are chosen.
The significance of genome duplication
Nothing is more fundamental to living organisms than the ability to reproduce. Each time a human cell divides, it must duplicate its genome once and only once; otherwise, the cell is programmed to commit suicide, an event termed apoptosis. For reproduction to occur, each cell selects from 20,000 to 40,000 sites throughout its 46 chromosomes at which it will initiate DNA replication. Selection of initiation sites is determined by the interaction between a DNA replication origin and a protein complex called the origin recognition complex (ORC). In addition, other proteins may facilitate this interaction and thereby contribute to initiation site selection.
Our goal is to discover how human cells determine where and when to initiate DNA replication and how the initiation process is regulated. During normal animal development, some cells are programmed to arrest mitosis and to duplicate their genomes many times in order to produce a large number of copies of critical genes without undergoing apoptosis. As we age, however, the ability to regulate DNA replication diminishes and results in genome instability and promiscuous cell division, the primary hallmarks of cancer. At least 122 human diseases result from aberrant DNA replication, mutation in DNA replication or repair genes, or infection by DNA viruses (DePamphilis, ed, DNA Replication and Human Disease. Cold Spring Harbor Press, 2006).
Several years ago, we discovered that the behavior of ORC in mammalian cells differs significantly from that in single-cell eukaryotes such as yeast. Yeast ORCs consist of a stable complex of six subunits that remain bound to chromatin throughout cell division and that target specific DNA sequences. In contrast, mammalian ORCs consist of a stable core complex of Orc2 through Orc5 that interacts weakly with Orc1 and Orc6. Nevertheless, the association of Orc1 with ORC(2-5) is essential for prereplication complex assembly and DNA replication. In vitro, however, metazoan ORCs exhibit little affinity for specific DNA sequences other than for asymmetric A:T-rich regions, whereas, in the differentiated cells of mammals and flies, ORCs localize at specific genomic sites that are coincident with DNA replication origins. Thus, the ability of ORC to activate a particular replication origin appears to depend on its ability to interact with DNA as it exists within the nucleus, an interaction that appears to be regulated by Orc1 (Figure 1).
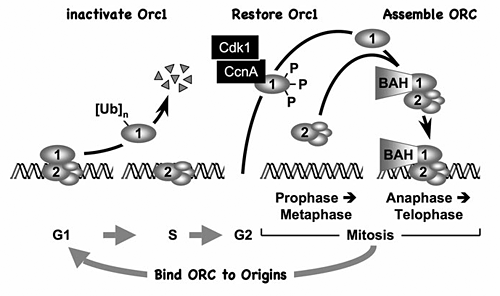
Figure 1. The ORC cycle in mammalian cells (reviewed in DePamphilis et al., Curr Opin Cell Biol 2006;18:231)
ORC is bound to chromatin during the G1 phase of the cell cycle, at which time it is part of a prereplication complex. When S phase begins, human Orc1 is selectively degraded by the 26S proteasome via a ubiquitin-dependent mechanism and reappears during the M- to G1-phase transition. During metaphase, Orc1 is hyperphosphorylated, an event that prevents ORC assembly. As cells exit metaphase, Orc1 is dephosphorylated and, together with other ORC subunits, binds to chromatin, an event that is facilitated by the Orc1 BAH domain.
During the past year, we established the basic features of the ORC cycle, which is a regulatory pathway that we previously proposed; it restricts initiation of DNA replication events so that genomes are duplicated once and only once each time a cell divides. Through its BAH domain, the largest subunit (Orc1) regulates association of the stable ORC(2-5) core complex with replication origins in vivo. The BAH domain interacts with another, as yet unidentified, protein that is required for ORC binding to chromosomes as cells exit mitosis and begin a new cell division cycle. Moreover, if Orc1 is not bound to ORC(2-5), it can induce apoptosis. However, if the unbound Orc1 is either monoubiquitinated or phosphorylated (two normal cell cycle–dependent modifications of Orc1), the modified Orc1 is localized in the cytoplasm where it cannot initiate replication, thus accounting for the fact that Orc1 is modified and in some cells degraded as the cells enter S phase (period of DNA replication). The modified forms of Orc1 do not induce apoptosis. Thus, cell cycle modifications regulate Orc1 activity, and Orc1 activity regulates ORC activity, which regulates initiation of DNA replication.
During mammalian development, genome endoreduplication is a rare event, and its mechanism is unknown. It first appears when FGF4 deprivation induces differentiation of trophoblast stem (TS) cells into the nonproliferating trophoblast giant (TG) cells required for embryo implantation. We discovered that selective inhibition of CDK1, the enzyme required to enter mitosis, by p57/Kip2 (a CDK-specific inhibitor in mammals) induced differentiation of TS cells into TG cells but induced abortive endoreduplication and apoptosis in embryonic stem cells. Therefore, inactivation of CDK1 triggers endoreduplication only in cells programmed to differentiate into polyploid cells. TS cell mutants revealed that p57 was required to trigger endoreduplication by inhibiting CDK1 while p21 (another CDK-specific inhibitor) suppressed expression of the checkpoint protein kinase CHK1, thereby preventing induction of apoptosis. Furthermore, Cdk2−/− TS cells revealed that CDK2 is required for endoreduplication when CDK1 is inhibited. Expression of p57 in TG cells was restricted to G-phase nuclei to allow CDK activation of S phase. Thus, endoreduplication in TS cells is triggered by p57 inhibition of CDK1 with concomitant suppression of the DNA damage response by p21.
The significance of early mammalian development
Mammalian development begins when an egg is fertilized by a sperm. This event activates the first round of genome duplication, after which the fertilized egg cleaves into a two-cell embryo. In mice (a laboratory model for human development), the two-cell embryo then activates the expression of about 300 genes that are required to continue development of the organism. Initially, each of the cells (blastomeres) produced by the cleavage events are “totipotent”; that is, they are capable of giving rise to the entire organism. But within five rounds of cell cleavage, a blastocyst appears, marking the beginning of cell differentiation (the process whereby cells become specialized). Blastocysts consist of a spherical monolayer of cells called the trophectoderm that gives rise to TS cells and eventually to the placenta. The trophectoderm layer encompasses a group of cells called the inner cell mass that gives rise to embryonic stem (ES) cells and eventually the embryo. Life begins after the blastocyst implants into the uterine endometrium and initiates formation of a nervous system.
Our goal is to identify genes whose expression is required for development to begin and then to identify the role played by these genes in establishing a living organism. We expect that such genes are essential for genome duplication, cell proliferation, and differentiation of the totipotent blastomeres into placental and embryonic cell lineages.
Genome duplication undergoes two dramatic changes during animal development. First, the number and location of replication origins can and do change at specific times during development. Second, the several concerted pathways that restrict genome duplication to once per cell division are inactivated at a few specific stages during development in order to allow several cycles of genome duplication to occur without an intervening mitosis. This process is called endoreduplication, and it occurs in mammals when TS cells differentiate into trophoblast giant cells during the process of embryo implantation.
During the past year, we identified the biological roles of three genes that are unique to vertebrates and that are expressed at the very beginning of mammalian development (Figure 2). We discovered all three genes and note that one, Dkkl1, is unique to mammals. We observed that Dkkl1 is expressed specifically during implantation of the embryo and development of spermatocytes into sperm. Moreover, genetic inactivation of DKKL1 in mice resulted in production of sperm that are defective in fertilization.
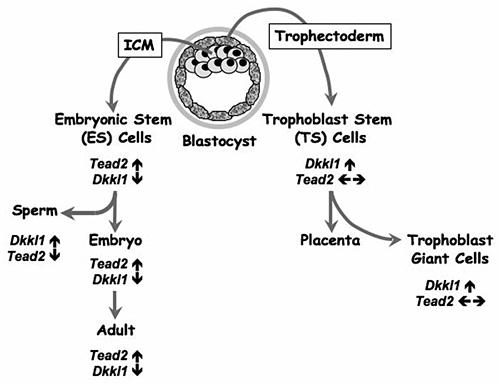
Figure 2. Differential expression of Tead2 and Dkkl1 mark divergent pathways to embryonic and placental tissues
The blastocyst is composed of two cell types—an outer layer of trophectodermal cells and the intermediate cell mass (ICM). The trophectoderm forms most of the extra-embryonic tissues and the placenta. Trophoblast stem (TS) cells may be derived from the trophectoderm cells that overlie the ICM, which gives rise to the embryo. Embryonic stem (ES) cells may be derived from the ICM. Northern blotting-hybridization, RT-PCR, and EST database analyses reveal that the trophectoderm and its derivatives strongly express Dkkl1 (↑), whereas Tead2 expression remains unchanged (← →). In contrast, the ICM and its derivatives strongly express Tead2 (↑) and suppress expression of Dkkl1 (↓). One notable exception occurs during the production of male germ cells; developing spermatocytes strongly express Dkkl1 in the absence of Tead2 expression such that this Dkkl1 is eventually localized to the acrosome of mature sperm.
The second gene, Tead2, is one of a highly conserved family of four transcription factors that share a common DNA binding domain. Surprisingly, TEAD2 is not required until after implantation has occurred and the nervous system begins to form. When we genetically inactivated TEAD2, mice had difficulty in forming a neural tube. In developing vertebrates, the neural tube is the embryo’s precursor to the central nervous system, which comprises the brain and spinal cord. Failure to close the neural tube in mice is called exencephaly, which is related to anencephaly, a common human birth defect that can be prevented by folic acid.
The third gene is Tead4. We discovered that Tead2 and Tead4 are the only Tead genes that are expressed in pre-implantation mouse embryos and that genetic inactivation of TEAD4 (but not of TEAD2) arrests development before formation of a blastocyst. In fact, we discovered that Tead4 is required for specification of the trophectoderm lineage, thus making it the earliest gene known so far on the pathway to formation of the placenta. It now appears that the totipotent blastomeres in mouse morula that express the transcription factor TEAD4 become trophectoderm and eventually placenta, whereas those that express the transcription factor OCT4 become the inner cell mass and eventually the embryo. Thus, Tead4 appears to be a master gene that sets into motion the first round of cell differentiation during mammalian development (Figure 3).
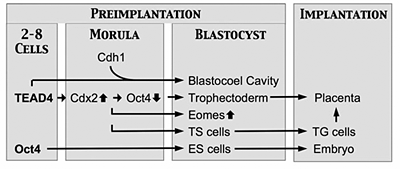
Figure 3.
Genes encoding transcription factors TEAD4 and OCT4 appear to function as master genes in that they mark the beginning of a chain of events that specifies a unique cell lineage. Oct4 is the first gene whose expression is required for specification of the embryonic cell lineage (ICM and ES cells) that produces the embryo. Tead4 is the first gene required for specification of the trophectoderm cell lineage that produces TS cells, trophoblast giant (TG) cells, and the placenta. In the absence of Tead4 expression, all totipotent blastomeres produce OCT4 protein. Expression of Tead4 induces expression of Cdx2, which suppresses expression of Oct4 and thus causes the blastomere to differentiate into trophectoderm instead of ICM.
Publications
- Ullah Z, Kohn MJ, Yagi R, Vassilev LT, DePamphilis ML. Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev 2008 22:3024-3036.
- Zhu W, DePamphilis ML. Selective killing of cancer cells by suppression of geminin activity. Cancer Res 2009 69:4870-4877.
- Kaneko KJ, Kohn MJ, Liu C, DePamphilis ML. The acrosomal protein Dickkopf-Like 1 (DKKL1) is not essential for fertility. Fertil Steril 2009 in press.
- Kohn MJ, Sztein J, Yagi R, DePamphilis ML, Kaneko KJ. The acrosomal protein Dickkopf-Like 1 (DKKL1) facilitates sperm penetration of the zona pellucida. Fertil Steril 2009 in press.
- Ullah Z, Lee CY, Lilly MA, DePamphilis ML. Developmentally programmed endoreduplication in animals. Cell Cycle 2009 8:1501-1509.
Collaborators
- James Inglese, PhD, NIH Chemical Genomics Center, Bethesda, MD
- Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Chengyu Liu, PhD, Transgenic Facility, NHLBI, Bethesda, MD
- Lyubomir Vassilev, PhD, Hoffmann-LaRoche, Nutley, NJ
- Jonathon Vogel, MD, Dermatology Branch, NCI, Bethesda, MD
- Yingming Zhao, PhD, University of Texas Southwestern Medical Center, Dallas, TX
Contact
For more information, email depamphm@mail.nih.gov or visit depamphilislab.nichd.nih.gov.