



You are here: Home > Section on Molecular Genetics of Immunity
Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Natarajan Ayithan, PhD, Visiting Fellow
- Tsung Hsien Chang, PhD, Visiting Fellow
- Anup Dey, PhD, Biologist
- Tomohiko Kanno, MD, PhD, Staff Scientist
- Mira Patel, PhD, Visiting Fellow
- Lakshmi Ramakrishna, PhD, Visiting Fellow
- Ryusuke Yoshimi, MD, PhD, Supplementary Visiting Fellow
We study transcription factors and chromatin binding proteins involved in innate immunity. One of the proteins we work on is IRF8, known to direct the development of dendritic cells (DCs) and macrophages. These cells provide resistance against pathogen infections. IRF8 and other related transcription factors are conjugated to ubiquitin and SUMO molecules in response to pathogen and interferon signals. We showed that TRIM21, an autoantigen present in patients with systemic lupus erythematosus, acts as a ubiquitin ligase for IRF8 and other IRF proteins to modulate their activity. To further study the role of TRIM21 in innate immunity, we knocked out the Trim21 gene in the mouse and studied the immunological phenotype. We found that the immune system in Trim21−/− mice is normal in gross anatomy and capable of mounting adaptive immune responses. However, Trim 21−/− cells showed hyperactivation of NFkB and produced excess proinflammatory cytokines in response to pathogens. These data indicate that TRIM21 is a negative regulator of inflammatory responses. We also showed that IRF7, critical for transcriptional induction of interferons (IFN), and related to IRF8, is SUMOylated after viral infection and inhibits IFN induction. Extending this work, we found that VP35, a viral anti-IFN protein derived from Ebola virus, amplifies IRF7 SUMO modification. It recruited proteins of the SUMOylation machinery to IRF7. The increased IRF7 SUMOylation led to reduced IFN induction, indicating that Ebola virus has a protein that inhibits host cell IFN induction to undermine innate immunity. We are also interested in the role of chromatin in innate immunity and investigate the bromodomain protein Brd4 and histone H3.3. Brd4 binds to acetylated chromatin in nuclear regions enriched with the variant histone H3.3. H3.3 is deposited onto DNA in association with transcription and is believed to play a role in epigenetic memory. With NIH3T3 cells expressing YFP–tagged H3.3, we examined incorporation of H3.3 in IFN–stimulated genes, and found that H3.3 is rapidly taken up by IFN–stimulated genes and was dramatically enriched in the 3′ coding regions. Surprisingly, H3.3 deposition continued long after IFN–stimulated transcription ceased. Furthermore, knockdown of endogenous H3.3 led to reduced transcription of IFN–stimulated genes, indicating that H3.3 deposition plays a role in regulating IFN–stimulated transcription and leaves a lasting epigenetic mark on IFN–stimulated genes.
Disruption of the Trim21 gene reveals its role in the negative regulation of Toll-like receptor–mediated NFκB activation
TRIM21 is a member of the large tripartite motif family, consisting of more than 60 members, many of which carry the RING-type zinc finger with a predicted ubiquitin ligase activity. One member, TRIM21, also known as Ro52, is an autoantigen found in patients with autoimmune diseases such as systemic lupus erythematosus. TRIM21 is an IFN–inducible protein expressed in many cell types. We previously reported that IRF8 is ubiquitinated by TRIM21 in macrophages after IFN stimulation, and that this ubiquitination augments the expression of IL-20p40 that requires IRF8. This result is significant, given that IL-12 controls many aspects of innate immunity. TRIM21 is also known to ubiquitinate IRF3, although the biological consequence of this ubiquitination has remained obscure. In collaboration with Herbert Morse’s laboratory, we disrupted the murine Trim21 gene by homologous recombination and replaced the locus with the sequence encoding the green fluorescent protein (GFP). This allowed us to study detailed expression of Trim21 mRNA in various cell types as well as Trim21's immunological functions. We showed that Trim21 is expressed in all immune cells, with the highest levels found in dendritic cells (DC), macrophages, and B lymphocytes, and is induced by IFNs. Trim21−/− mice were found to be fertile and to show no gross abnormality in the anatomical structure of immune organs as well as in the make up of immune cells. DCs and macrophages responded normally to pathogen signals through the toll-like receptors (TLR). Close examinations of fibroblasts, however, showed that, in response to typical TLR signals such as LPS and Poly IC, NFκB is activated at higher levels in Trim21−/− cells than in the wild-type cells. As a consequence, Trim 21−/− cells overproduced several proinflammatory cytokines, specifically IL-1 and IL-6. These data indicated that TRIM21 regulates excessive inflammatory responses by acting on the NFκB activation process. Further observations demonstrated that TRIM21 is capable of ubiquitinating TRAF6, an adaptor that plays a critical role in NFκB activation. Trim21 maps to a region of chromosome 6 that also harbors a number of similar Trim genes. We found that the expression of other Trim genes is up-regulated in Trim21−/− cells, indicating a compensatory mechanism created by the network of crosstalk among Trim family proteins.
The role of Brd4 and histone H3.3 deposition in interferon-induced transcription
Brd4, the conserved bromodomain protein, binds to acetylated histones H3 and H4 both in vivo and in vitro. In addition, Brd4 remains associated with chromatin during mitosis and meiosis, a feature unusual for chromatin- and DNA-binding proteins. A structurally related protein, Brd2 also shares this feature. Thus, Brd4 and Brd2 are implicated in epigenetic memory. We previously showed that Brd4 interacts with an elongation factor P-TEFb, composed of cyclin T1 and Cdk9 and participates in transcription of HIV and many cellular genes. To gain insight into the role of Brd4 in transcriptional memory, we performed chromatin immunoprecipitation (ChIP) analysis and found that Brd4 binds preferentially to the promoter region of many expressed genes where H3 and H4 are acetylated. We also showed that NFκb activation is associated with the recruitment of Brd4 to the proinflammatory cytokine genes and their transcriptional induction. In the past year, we extended our study to address the involvement of the histone variant H3.3 in Brd4–mediated transcription. Among core histones, the histone H3.3 has a distinct characteristic: while most core histones—H2A, H2B, H3.1, H3.2, and H4—are synthesized strictly during S phase and immediately deposited onto newly synthesized DNA, H3.3 is synthesized outside S phase and is believed to be deposited on actively expressed genes in a transcription-coupled manner. Accordingly, H3.3 has markers of post-translational modifications associated with gene activation, including H3 and H4 acetylation. Previously studies on H3. 3 behavior replied heavily on the Drosophila models and other non-mammalian systems. These studies indicated that H3.3 is enriched in the promoter regions of many active genes, suggesting that it has a role in transcriptional activation. In an effort to understand the activity of H3.3 in inducible gene expression in the mammalian system, we examined deposition of H3.3 in IFN–stimulated genes in NIH 3T3 cells. Using NIH3T3 cells expressing H3.3 tagged with Yellow fluorescent protein (YFP), we tested their deposition in a number of IFN responsive genes by chromatin immunoprecipitation (ChIP) analysis. Our results showed that H3.3 is deposited rapidly in all IFN responsive genes tested after stimulation. The most extensive deposition was noted in the 3’ end of the coding region of these genes. IFN responsive genes were transcribed rapidly after stimulation, peaking at around 6 hours followed by complete cessation of transcription in 24 hours. Surprisingly, H3.3 deposition in these genes continued for at least 24 hours following the completion of transcription. Further, contrary to expectation, H3.3 did not show an increased deposition at the promoter region. The pattern and kinetics of H3.3 deposition was somewhat similar to that of H3K36 trimethylation, as this modification was induced by IFN which continued even after IFN stimulation. However H3.3 deposition did not correlate with changes in histone acetylation. These results indicated that H3.3 deposition is coupled with transcriptional activation and has a long-term consequence for the chromatin status of IFN stimulated genes.
Ebola virus undermines innate immunity by hijacking the host SUMOylation machinery: the role of the viral VP35 protein in IRF7 SUMO modification
Ebola is an acute hemorrhagic disease that kills the large majority of infected individuals and is caused an RNA virus. Although outbreaks have been seen only in Africa, Ebola virus, because of its potential public health threat in this and other countries, is classified as a Category A bioterrorism agent. The virus initially infects macrophages and DCs and weakens innate immune functions through the viral protein VP35. By studying the mouse model of Ebola virus infection, we found that VP35 inhibits the activity of IRF7, the transcription factor that initiates IFN transcription in DCs. Yeast two-hybrid screens and co-immunoprecipitation analyses identified UBC9, PIAS1, and IRF7 as proteins that interact with VP35 in activated DCs. UBC9 and PIAS1 are, respectively, an E2 enzyme and E3 ligase in the SUMO conjugation reaction. We found that IRF7 SUMO modification was markedly increased in the presence of VP35 as a consequence of the increased interaction of IRF7 with PIAS1. In accordance with these results, PIAS1, but not VP35, was found to act as a SUMO ligase for IRF7. In the presence of VP35, IRF7 was SUMOylated not only at the major SUMO site K406 but on an additional non-canonical lysine residue. We also found that VP35 and PIAS1 together strongly inhibited IFN promoter activity. Supporting combined activities of PIAS1 and VP35, sh RNA for PIAS1 significantly reduced the inhibition of IRF7 dependent IFN transcription. These results illustrate how the Ebola virus VP35 hijacks the SUMO conjugation system to prematurely downregulate IFN transcription in DCs, thereby increasing viral propagation in infected individuals.
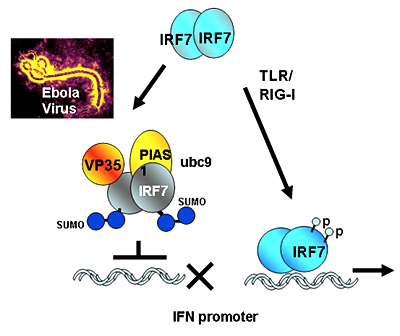
Figure 1. The Ebola virus protein VP35 hijacks the SUMO conjugation machinery and weakens innate immunity.
Pathogen signals activate IRF7 to stimulate interferon transcription (right) in Ebola virus infection (left), the viral protein VP35 binds to UBC9 and PIAS1, the host SUMO E2 and E3 enzymes, respectively, and brings them to IRF7, causing premature inhibition of IFN transcription.
Additional Funding
- Trans NIH-FDA Biodefense Program, IATAP Program, I-to-I Program
Publications
- Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M, Kato A, Ozato K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem 2008 283:25660-25670.
- Ozato K, Shin DM, Chang TH, Morse HC. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 2008 8:849-860.
- Kim JY, Ozato K. The sequestosome 1/p62 attenuates cytokine gene expression in activated macrophages by inhibiting IFN regulatory factor 8 and TNF receptor-associated factor 6/NF-kappaB activity. J Immunol 2009 182:2131-2140.
- Tamura T, Smith M, Kanno T, Dasenbrock H, Nishiyama A, Ozato K. Inducible deposition of histone variant H3.3 in interferon stimulated genes. J Biol Chem 2008 283:12217-12225.
- Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathogens 2009 6:e1000493.
Collaborators
- Toru Kubota, PhD, Japan National Institute of Infectious Diseases, Tokyo, Japan
- Ben-Zion Levi, PhD, Technion, Israel Institute of Technology, Haifa, Israel
- James McNally, PhD, Laboratory of Receptor Biology and Gene Expression, NCI, Bethesda, MD
- Herbert Morse, II, MD, Laboratory of Immunopathology, NIAI D, Rockville, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
Contact
For more information, email ozatok@mail.nih.gov or visit ozatolab.nichd.nih.gov.