

You are here: Home > Section on Drosophila Gene Regulation
Genomics of Development in Drosophila

- James A. Kennison, PhD, Head, Section on Drosophila Gene Regulation
- Monica T. Cooper, BA, Senior Research Technician
- Anwar Ogunsanya, BA, Postbaccalaureate Fellow
Our goal is to understand how linear information encoded in genomic DNA functions to control cell fates during development. The Drosophila genome is about twenty times smaller than the human genome. However, despite its smaller size, most developmental genes and at least half of the disease- and cancer-causing genes in man are conserved in Drosophila, making Drosophila a particularly important model system for the study of human development and disease. One of the important groups of conserved developmental genes are the homeotic genes. In Drosophila, the homeotic genes specify cell identities at both the embryonic and adult stages. The genes encode homeodomain-containing transcription factors that control cell fates by regulating the transcription of downstream target genes. The homeotic genes are expressed in precise spatial patterns that are crucial for the proper determination of cell fate. Both loss of expression and ectopic expression in the wrong tissues lead to changes in cell fate. The changes provide powerful assays for identifying the trans-acting factors that regulate the homeotic genes and the cis-acting sequences through which they act. The trans-acting factors are also conserved between Drosophila and human and have important functions, not only in development but also in stem-cell maintenance and cancer.
Cis-acting sequences for transcriptional regulation of the Sex combs reduced homeotic gene
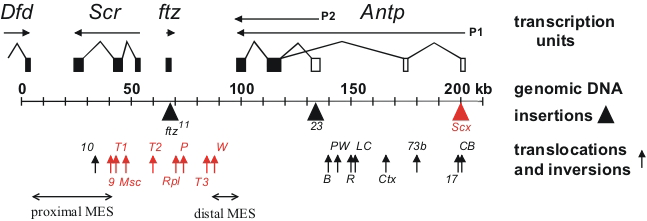
Click image to enlarge.
Figure 1. Chromosomal aberrations in the distal half of the Antennapedia complex
The transcription units are shown above the genomic DNA, while chromosomal aberrations are shown below (solid triangles indicate insertions of transposable elements and upward arrows indicate breakpoints of translocations and inversions). Chromosomal aberrations (shown in red) interfere with silencing in the adult second and third legs. The regions that include the proximal and distal MES are indicated by horizontal arrows.
Assays in transgenes in Drosophila previously identified cis-acting transcriptional regulatory elements from homeotic genes. The assays identified tissue-specific enhancer elements as well as cis-regulatory elements that are required for the maintenance of activation or repression throughout development. While these transgenic assays have been important in defining the structure of the cis-regulatory elements and identifying trans-acting factors that bind to them, their functions within the context of the endogenous genes remain poorly understood. We used a large number of existing chromosomal aberrations in the Sex combs reduced homeotic gene to investigate the functions of the cis-acting elements within the endogenous gene. The chromosomal aberrations identified an imaginal leg enhancer about 35 kb upstream of the Sex combs reduced promoter. The enhancer is not only able to activate transcription of the Sex combs reduced promoter that is 35 kb distant but can also activate transcription of the Sex combs reduced promoter on the homologous chromosome. Although the imaginal leg enhancer can activate transcription in all three pairs of legs, it is normally silenced in the second and third pairs of legs. The silencing requires the Polycomb-group proteins. We are currently attempting to identify the cis-regulatory DNA sequences in the Sex combs reduced gene that are required for Polycomb-group silencing in the second and third legs. Characterization of the chromosomal rearrangements shown in Figure 1 also revealed that two genetic elements (proximal and distal MES), about 70 kb apart in the Sex combs reduced gene, must be in cis to maintain proper repression. When not physically linked, the elements interact with elements on the homologous chromosome and cause derepression of its wild-type Sex combs reduced gene. Using a transgenic assay, we identified at least five DNA fragments from the Sex combs reduced gene that silence transcription from a reporter gene. The transcriptional silencers are clustered in the two regions whose interactions are required for the maintenance of silencing in the endogenous genes. We also use the silencer elements to screen for mutations in trans-acting silencing factors.
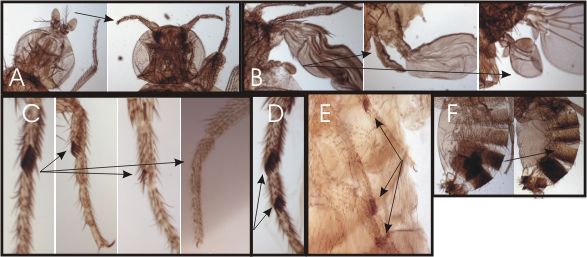
Click image to enlarge.
Figure 2. Homeotic phenotypes of new pharate-adult lethal mutants
(A) Wild-type on the left and the transformation of aristae to distal leg on the right. (B) Wild-type haltere on the left and transformations of anterior and posterior haltere to anterior and posterior wing in the middle and right, respectively. (C) First legs from a wild-type male on the left and three different mutants with reduced sex combs on the right. (D) Mutant male with sex combs on both the first and second tarsal segments. (E) Mutant male with sex combs on all three pairs of legs. (F) Abdominal segments from a wild-type male on the left; mutant male with transformation of the fifth abdominal segment to a more anterior identity on the right.
Trans-acting activators and repressors of homeotic genes
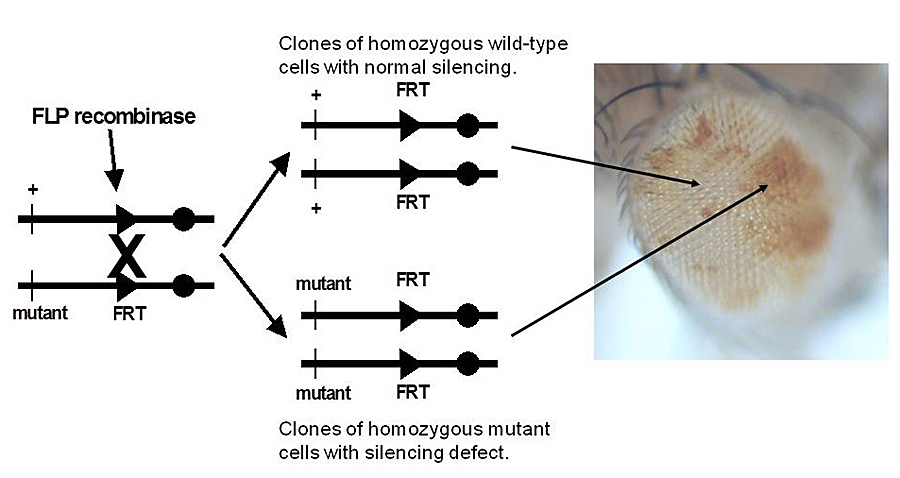
Click image to enlarge.
Figure 3. Genetic screen for new mutations that disrupt pairing-sensitive silencing
Flies homozgous for transposons carrying the mini-white reporter gene and a pairing-sensitive silencing element have white eyes. Clones of cells homozygous for newly induced mutants are generated using the yeast site-specific recombinase (FLP recombinase) and its target site (FRT). The clones of mutant cells are able to express the mini-white reporter gene and are pigmented (shown in the eye on the right of the figure).

Click image to enlarge.
Figure 4. New mutations that disrupt pairing-sensitive silencing
The eye in the top left is the control with normal pairing-sensitive silencing. The remaining eyes are from flies with new mutations that disrupt pairing-sensitive silencing. The homozygous mutant cells are able to express the mini-white reporter gene in the transposon and are pigmented.
The initial domains of homeotic gene repression are set by the segmentation proteins, which also divide the embryo into segments. Genetic studies identified the trithorax group of genes that are required for expression or function (such as maintenance of transcriptional activation) of the homeotic genes. Maintenance of transcriptional repression requires the proteins encoded by the Polycomb-group genes. To identify new trithorax-group activators and Polycomb-group repressors, we screened for new mutations that mimicked the following phenotypes: loss of function or ectopic expression of the homeotic genes. We generated over 4,000 lethal mutants and, among those that die late in development, identified two dozen mutants with homeotic phenotypes. Some of the homeotic phenotypes are shown in Figure 2. The mutants identify genes required for expression or function of the homeotic genes.
We also use pairing-sensitive silencing elements from the Sex combs reduced gene to screen for recessive mutations that interfere with homeotic gene silencing. In flies homozygous for transgenes with pairing-sensitive silencing elements, we generate clones of cells in the eye that are homozygous for newly induced mutations, using the yeast FLP/FRT site-specific recombination system (Figure 3). Silencing mutations are detected by the appearance of pigmented spots in the white-eyed flies. Several examples of the new mutations recovered are shown in Figure 4. About half the mutations recovered to date are in known Polycomb-group genes. The remaining mutations identify new genes required for silencing. The second-chromosome genes identified in the screen are shown in Figure 5; seven are known Polycomb-group genes. For ten of the fifteen new genes, the corresponding transcription units were identified by a combination of meiotic recombination mapping and whole-genome sequencing.
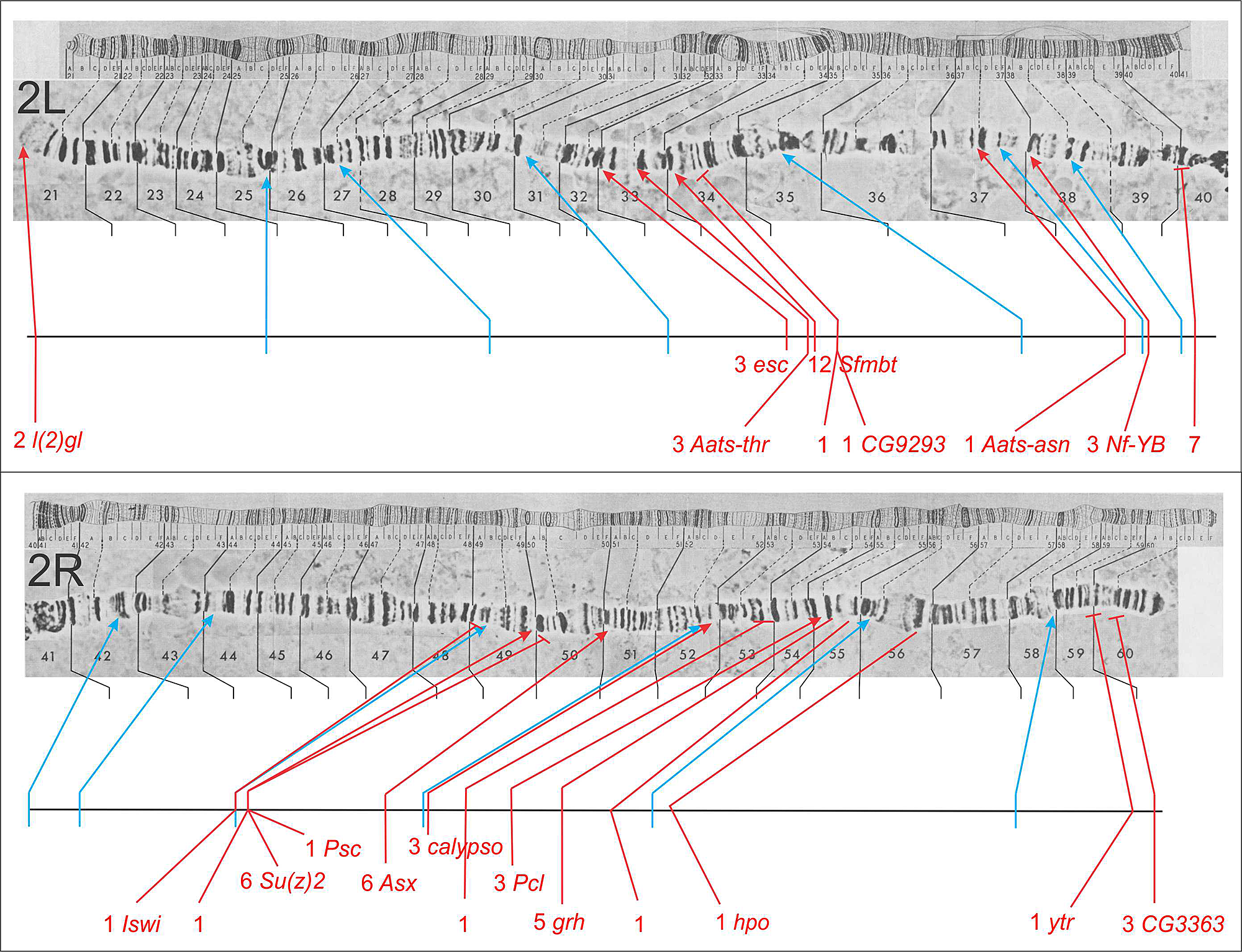
Click image to enlarge.
Figure 5. Genes identified by second chromosome mutants defective for pairing-sensitive silencing
The top half of the figure is the left arm of the second chromosome (2L) and the bottom half is the right arm (2R). The genes identified by mutants in the screen for defects in pairing-sensitive silencing are shown below the polytene salivary gland chromosomes. For each gene, the number of alleles recovered is indicated, with a red line showing the location of the gene on the chromosome map. Blue lines indicate the locations of the transposon insertions used to initially map the mutants by meiotic recombination. Following the number of alleles for a gene is the gene name (if the transcription unit corresponding to the gene has been identified).
Structure and function of the Drosophila genome
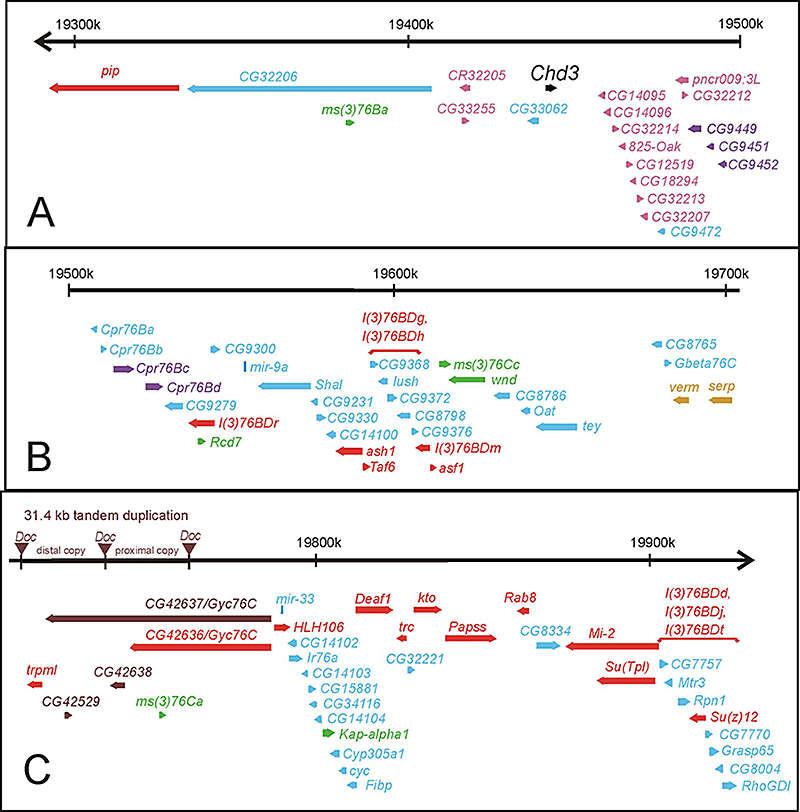
Click image to enlarge.
Figure 6. Molecular map of the genomic region deleted in Df(3L)kto2 (polytene region 76B-D)
The approximately 640 kb of genomic DNA (from 3L: 19291k to 19926k, Release 5.23) is broken into three parts (A, B, and C) and is represented by the horizontal black arrows at the top of each part. The annotated transcription units are represented by colored thick horizontal arrows. Transcription units essential for viability are red and gold. Transcription units essential only for fertility are green. The clusters of transcription units encoding related proteins are purple, pink (the OAK cluster in 76B), and gold (the verm and serp genes essential for viability). The non-essential transcription unit Chd3 is in black. All other transcription units are blue. The two regions that include the five essential gene for which the transcription units have not been identified [l(3)76BDg, l(3)76BDh, l(3)76BDd, l(3)76BDj, and l(3)76BDt] are indicated by red horizontal brackets above the candidate transcription units. A 31.4 kb tandem duplication (distal copy and proximal copy) flanked by Doc transposable elements in the sequenced iso-1 strain (but not in other wild-type strains) is shown on the genomic DNA at the left of Panel C, with the Doc elements represented by inverted brown triangles.
The Drosophila melanogaster genome has been intensely studied for over 100 years. Recently, sequencing of the majority of the genomic DNA revealed much about the structure and organization of the genome. Despite the molecular advances, much remains to be discovered about the functions encoded within the genome. As part of a long-term project to understand the function and organization of the Drosophila genome, we set out to identify all genes essential for viability or male fertility in two regions of the genome that span almost 1 megabases of DNA (shown in Figures 6 and 7). We identified 10 gene clusters that appear to have arisen by tandem duplication. The clusters include 34 of the 137 predicted genes. We identified 47 genes essential for zygotic viability, including two of the pairs of tandemly duplicated genes. We also identified seven genes that are required only for fertility, most of which are male-specific in expression. Our analysis of data from the modENCODE project suggests that 20% or more of the genome is expressed only in males. We were also able to assess the progress of the Drosophila Gene Disruption Project. While the project has tagged about two-thirds of the annotated genes with transposon insertions, these insertions only disrupt the function of 45% of the genes.
Surprisingly, when we deleted a genomic region that spanned 55 kb, we found no effects on either viability or fertility. Although this nonessential region includes seven predicted genes, there is no evidence that the genes are expressed under any known conditions. The seven predicted genes are also not evolutionarily conserved. While there are no evolutionarily conserved open reading frames within this gene desert, the region has 48 DNA sequences of between 12 and 33 base pairs each that are identical in 12 different Drosophila species. The strong conservation indicates that the sequences must have some function that is beneficial to flies living outside the laboratory.
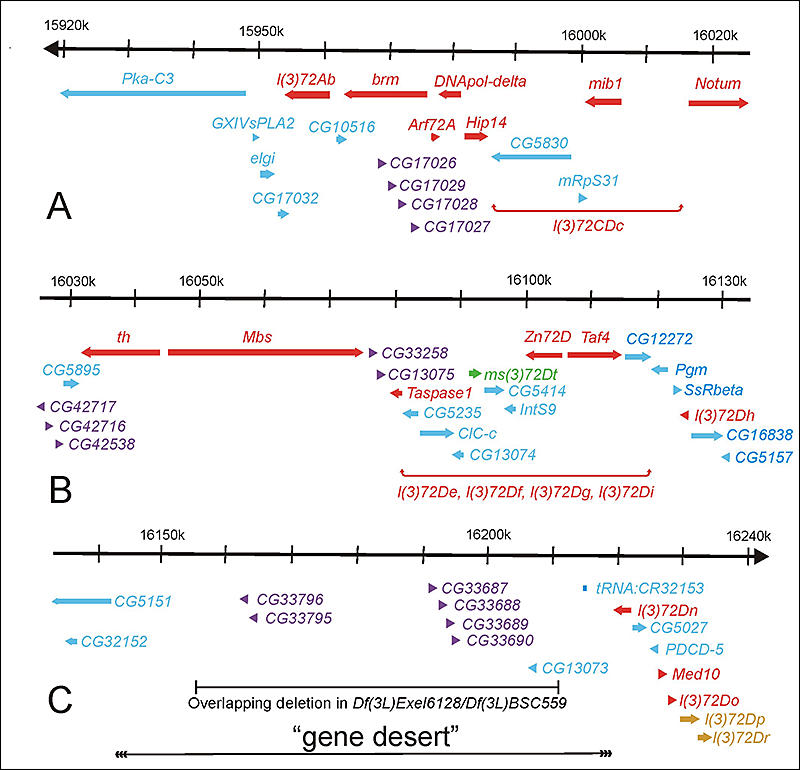
Click image to enlarge.
Figure 7. Molecular map of the genomic region deleted in Df(3L)th102 (polytene region 72A-D)
The approximately 320 kb of genomic DNA (from 3L: 15918k to 16240k, Release 5.23) is broken into three parts (A, B, and C) and is represented by the horizontal black arrows at the top of each part. The annotated transcription units are represented by colored thick horizontal arrows. Transcription units essential for viability are red and gold. Transcription units essential only for fertility are green. The clusters of transcription units encoding related proteins are purple and gold [the l(3)72Dp and l(3)72Dr genes essential for viability]. All other transcription units are blue. The two regions that include the five essential genes for which the transcription units have not been identified [l(3)72CDc, l(3)72De, l(3)72Df, l(3)72Dg, and l(3)72Di] are indicated by red horizontal brackets below the candidate transcription units. The DNA missing in flies trans-heterozygous for the overlapping deletions Df(3L)Exel6128 and Df(3L)BSC559 and the putative “gene desert” are indicated by the horizontal black bars at the bottom of C.
Publications
- Stultz BG, Park SY, Mortin MA, Kennison JA, Hursh DA. HOX proteins coordinate peripodial decapentaplegic expression to direct adult head morphogenesis in Drosophila. Dev Biol 2012;369:362-376.
- Monribot-Villanueva J, Juarez-Uribe RA, Palomera-Sanchez Z, Gutierrez-Aguiar L, Zurita M, Kennison JA, Vazquez M. TnaA, an SP-RING protein, interacts with Osa, a subunit of the chromatin remodeling complex BRAHMA and with the SUMO pathway in Drosophila melanogaster. PLoS One 2013;8:e62251.
- Cunningham MD, Gause M, Cheng Y, Noyes A, Dorsett D, Kennison JA, Kassis JA. Wapl antagonizes cohesin binding and promotes Polycomb-group silencing in Drosophila. Development 2012;139:4172-4179.
- Lindsley DL, Roote J, Kennison JA. Anent the genomics of spermtogenesis in Drosophila melanogaster. PLos One 2013;8:e55915.
Collaborators
- Dale Dorsett, PhD, St. Louis University, St. Louis, MO
- Der-Hwa Huang, PhD, Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan
- Deborah A. Hursh, PhD, Center for Biologics Evaluation and Research, FDA, Bethesda, MD
- Judy Kassis, PhD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
- Dan Lindsley, PhD, University of California San Diego, La Jolla, CA
- Martha Vazquez, PhD, Instituto de Biotecnología, UNAM, Cuernavaca, Mexico
- Mario Zurita, PhD, Instituto de Biotecnología, UNAM, Cuernavaca, Mexico
Contact
For more information, email kennisoj@mail.nih.gov.