

You are here: Home > Section on Genomic Imprinting
Molecular Genetics of an Imprinted Gene Cluster on Mouse Distal Chromosome 7

- Karl Pfeifer, PhD, Head, Section on Genomic Imprinting
- Claudia Gebert, PhD, Research Fellow
- Bokkee Eun, PhD, Visiting Fellow
- Megan Sampley, PhD, Postdoctoral Fellow
- Barbara Pelham-Webb, BS, Postbaccalaureate Fellow
- Matthew Van Winkle, BA, Postbaccalaureate Fellow
- Christopher Yoon, BS, Postbaccalaureate Fellow
Genomic imprinting is an unusual form of gene regulation by which an allele’s parental origin restricts allele expression. For example, almost all expression of the non-coding RNA tumor suppressor H19 gene is from the maternal chromosome. In contrast, expression of the neighboring insulin-like growth factor 2 gene (Igf2) is from the paternal chromosome. Imprinted genes are not randomly scattered throughout the chromosome but rather are localized in discrete clusters. One cluster of imprinted genes is located on the distal end of mouse chromosome 7 (Figure 1). The syntenic region in humans (11p15.5) is highly conserved in gene organization and expression patterns. Mutations disrupting the normal patterns of imprinting at the human locus are associated with developmental disorders and many types of tumors including Wilms' Tumor and rhabdosarcomas in children. In addition, inherited cardiac arrhythmia is associated with mutations in the maternal-specific Kcnq1 gene. We use mouse models to address the molecular basis for allele-specific expression in this distal 7 cluster. We use imprinting as a tool to understand the fundamental features of epigenetic regulation of gene expression. We also generate mouse models for the several inherited disorders of humans, specifically models to study defects in cardiac repolarization associated with loss of Kcnq1 function, and recently we characterized the phenotype associated with loss of Calsequestrin2 gene function.
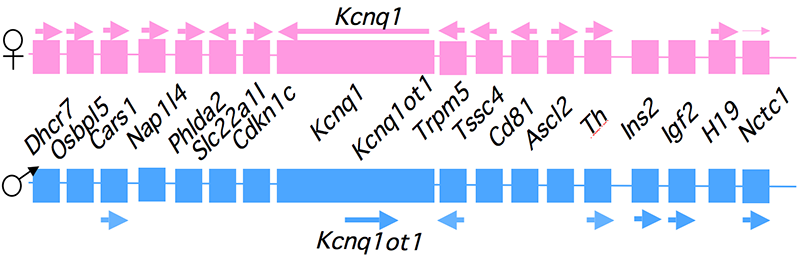
Click image to enlarge.
Figure 1. An imprinted domain on mouse distal chromosome 7
Maternal (pink) and paternal (blue) chromosomes are indicated. Horizontal arrows denote RNA transcription.
Alternative long-range interactions between distal regulatory elements establish allele-specific expression at the Igf2/H19 locus.
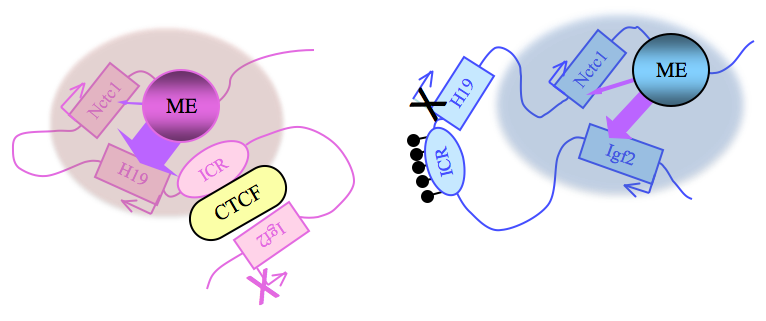
Click image to enlarge.
Figure 2. Distinct maternal and paternal chromosomal conformations at the distal 7 locus
Epigenetic modifications on the 2.4 kb ICR generate alternative 3D organizations across a large domain on paternal (blue) and maternal (pink) chromosomes and thereby regulate gene expression. ICR, imprinting control region; EE, endodermal enhancer; ME, muscle enhancer; filled lollipops covering the paternal ICR, CpG methylation.
Our studies on the mechanisms of genomic imprinting focus on the H19 and Igf2 genes, which lie at one end of the distal 7 imprinted cluster (Figure 1). Paternally expressed Igf2 lies about 80 kb upstream of the maternal-specific H19 gene. Using cell culture systems as well as transgene and knockout experiments in vivo, we identified the enhancer elements responsible for activation of these two genes. The elements are shared and are all located downstream of the H19 gene (Figure 2).
Imprinting at the Igf2/H19 locus is dependent upon the 2.4 kb H19 Imprinting Control Region (H19ICR) that lies between the two genes, just upstream of the H19 promoter (Figure 2). On the maternal chromosome, binding of the CTCF protein to the ICR establishes a transcriptional insulator that organizes the chromosome into loops that favor H19 expression but block interactions between the maternal Igf2 promoters and the downstream shared enhancers, thus preventing maternal Igf2 expression. Upon paternal inheritance, the CpG sites within the ICR are methylated, which prevents binding of the CTCF protein so that a transcriptional insulator is not established. Thus, paternal Igf2 promoters and the shared enhancers do interact via DNA loops, and expression of paternal Igf2 is facilitated. Altogether, we find that the fundamental role of the ICR is to organize the chromosomes into alternative 3-D configurations that promote or prevent expression of the Igf2 and H19 genes.
The H19ICR is not only necessary but is also sufficient for genomic imprinting. To demonstrate this, we used knock-in experiments to insert the 2.4 kb element at heterologous loci and demonstrated its ability to imprint these regions (1). Further, analyses of these loci confirmed and extended the transcriptional model described above. Upon maternal inheritance, even ectopic ICR elements remain unmethylated, bind to CTCF protein, and form transcriptional insulators. Paternally inherited ectopic ICRs become methylated, cannot bind to CTCF, and therefore promote alternative loop domains distinct from those organized on maternal chromosomes. Most curious was the finding that DNA methylation of ectopic ICRs is not acquired until relatively late in development, after the embryo implants into the uterus. (In contrast, at the endogenous locus, ICR methylation occurs during spermatogenesis.) These findings thus imply that DNA methylation is not the primary imprinting mark that distinguishes maternally from paternally inherited ICRs. Our current studies focus on identifying this true imprinting mark.
The long non-coding RNA Nctc1 lies downstream of H19 and is transcribed across the muscle enhancer element (ME in Figure 2) that is shared by Igf2 and H19. Nctc1 expression depends on this enhancer element. Concordantly, the shared enhancer interacts with the Nctc1 promoter just as it interacts with the maternal H19 and the paternal Igf2 promoters. We showed that all three co-regulated promoters (Igf2, H19, and Nctc1) also physically interact with each other in a manner that depends on their interactions with the shared enhancer. Thus, enhancer interactions with one promoter do no preclude interactions with another promoter. Moreover, we demonstrated that these promoter-promoter interactions are regulatory, and they explain the developmentally regulated imprinting of Nctc1 transcription. Taken together, our results demonstrate the importance of long-range enhancer-promoter and promoter-promoter interactions in physically organizing the genome and establishing the gene expression patterns that are crucial for normal mammalian development (2).
Molecular mechanisms for tissue-specific promoter activation by distal enhancers
Normal mammalian development is absolutely dependent on establishing the appropriate patterns of expression of thousands of developmentally regulated genes. Most often, development-specific expression depends on promoter activation by distal enhancer elements. The Igf2/H19 locus is a highly useful model system for investigating mechanisms of enhancer activation. First, the biological significance of the model is clear, given that expression of these genes is so strictly regulated. Even two-fold changes in RNA levels are associated with developmental disorders and with cancer. Second, we already know much about the enhancers in this region and have established powerful genetic tools to investigate their function. Igf2 and H19 are co-expressed throughout embryonic development and depend on a series of tissue-specific enhancers that lie between 8 and more than 150 kb downstream of the H19 promoter (or between 88 and more than 130 kb downstream of the Igf2 promoters). The endodermal and muscle enhancers have been precisely defined, and we generated mouse strains carrying deletions that completely abrogate enhancer function. We also generated insulator insertion mutations that specifically block muscle enhancer activity. We used these strains to generate primary myoblast cell lines so that we can combine genetic, molecular, biochemical, and genomic analyses to understand the molecular bases for enhancer functions.
A long non-coding RNA is an essential element of the muscle enhancer (3).
Transient transfection analyses define a 300–bp element that is both necessary and sufficient for maximal enhancer activity. However, stable transfection and mouse mutations indicate that this core element is not sufficient for enhancer function in a chromosomal context. Instead, the Nctc1 promoter element is also essential. (The Nctc1 gene encodes a spliced, polyadenylated long non-coding RNA). The Nctc1 RNA itself is not required (at least in trans). Instead mutational analysis demonstrates that it is Nctc1 transcription through the core enhancer that is necessary for enhancer function. Curiously, the Nctc1 promoter has chromatin features typical of both a classic enhancer and a classic peptide-encoding promoter. Several recent genomic studies have also suggested a role for non-coding RNAs in gene regulation and enhancer function. We will use our model system to characterize the role of Nctc1 transcription in establishing enhancer orientation, enhancer promoter specificity, and enhancer tissue specificity.
The muscle enhancer (ME) directs RNA polymerase II (RNAP) not only to its cognate promoters (i.e., to the H19 and Igf2 promoters) but also across the entire intergenic region.
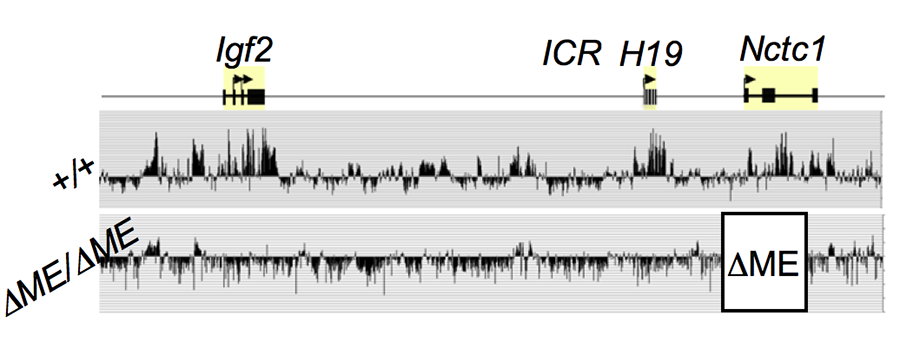
Click image to enlarge.
Figure 3. The shared muscle enhancer (ME) directs RNAP binding and RNA transcription across the entire 150 kb locus.
We used ChIP-on-chip to analyze RNAP localization on chromatin prepared from wild-type and from enhancer-deletion (ΔME) cell lines (Figure 3). As expected, RNAP binding to the H19 and Igf2 promoters is entirely enhancer-dependent. Curiously, we also noted enhancer-dependent RNAP localization across the entire locus including the large intergenic domain between the two genes. Furthermore, this RNAP binding is associated with RNA transcription. Thus, the enhancer regulates accessibility and RNAP binding not only at specific localized sites but across the entire domain. The results support a facilitated tracking model for enhancer activity.
RNAP binding at "real" genes and across the intergenic regions is qualitatively different.
We used naturally occuringr SNPs to investigate allelic differences in binding of RNAP and activation of gene expression in wild-type cells and in cells carrying enhancer deletions or insulator insertion mutations. RNAP binding across the Igf2 and H19 genes is both enhancer-dependent and insulator-sensitive; that is, a functional insulator located between an enhancer and its regulated gene prevents RNAP binding and likewise prevents RNA transcription. Across the intergenic regions, RNAP binding and RNA transcription are likewise enhancer-dependent (see above). However, such intergenic RNAP binding and transcripiton are not insulator-sensitive. The results indicate that insulators do not serve solely as a physical block for RNAP progression but rather they specifically interfere with certain RNAP states or activities.
Role of calsequestrin2 in regulating cardiac function
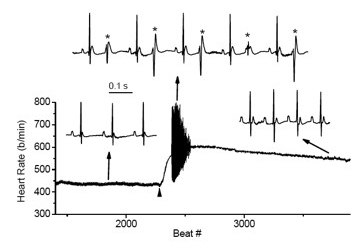
Figure 4. Cardiac arrhythmias in calsequestrin-2–deficient mice phenocopy the human disease.
Premature ventricular complexes (*) are induced by stress in Casq2–deficient but not in wild-type mice.
Mutations in the CASQ2 gene, which encodes cardiac calsequestrin (CASQ2), are associated with catecholaminergic polymorphic ventricular tachycardia (CPVT) and sudden death. The survival of individuals homozygous for loss-of-function mutations in CASQ2 is surprising, given the central role of Ca2+ ions in excitation-contraction (EC) coupling and the presumed critical roles of CASQ2 in regulating Ca2+ release from the sarcoplasmic reticulum (SR) into the cytoplasm. To address this paradox, we generated a mouse model for loss of Casq2 gene activity. Comprehensive analysis of cardiac function and structure generated several important insights into CASQ2 function (4). First, CASQ2 is not essential to provide sufficient Ca2+ storage in the SR of the cardiomyocyte. Rather, a compensatory increase in SR volume and surface area in mutant mice appears to maintain normal Ca2+ storage capacity. Second, CASQ2 is not required for the rapid, triggered release of Ca2+ from the SR during cardiomyocyte contraction. Rather, the RyR receptor opens appropriately, resulting in normal, rapid flow of Ca2+ into the cytoplasm, thus allowing normal contraction of the cardiomyocyte. Third, CASQ2 is required for normal function of the RyR during cardiomyocyte relaxation. In the absence of CASQ2, significant Ca2+ leaks occur through the RyR and lead to premature contractions and cardiac arrhythmias (Figure 4). Fourth, CASQ2 function is required to maintain normal junctin and triadin levels. We do not yet understand what role, if any, the compensatory changes in these two SR proteins play in modulating the loss of Casq2 phenotype.
To address these issues and to model cardiac disorders associated with late-onset (not congenital) loss of CASQ2 activity, we established and are analyzing two new mouse models in which changes in Casq2 gene structure are induced by hormone treatment. In the first model, an invested/null allele is restored to normal function by the addition of hormone. In the past year, we demonstrated the effectiveness of this model and noted that full Casq2 protein levels are restored within one week of treatment. In the second model, a functional gene is ablated by the addition of hormone. The Casq2 gene and mRNAs are deleted from cardiac cells within four days of hormone treatment. Phenotypic analyses shows that the mice present a profoundly more defective heart than congenitally mutant mice; that is, arrhythmias are more frequent, of longer duration, and qualitatively much more severe. Our results indicate that the developmental program has flexibility in coping with the loss of Casq2 protein that the adult heart lacks. Our current efforts focus on finding the molecular basis for the coping mechanisms as a means to better understand cardiac development and to perhaps identify clues as to how to handle Casq2 deficiency, clues that may be of therapeutic value.
Publications
- Shrimali S, Srivastava S, Varma G, Grinberg A, Pfeifer K, Srivastava M. An ectopic CTCF dependent transcriptional insulator influences the choice of Vβ gene segments on VDJ recombination at the TCRβ locus. Nucleic Acids Res 2012;40:7753-7765.
- Eun B, Sampley ML, Good AL, Gebert CM, Pfeifer K. Promoter cross-talk via a shared enhancer explains paternally biased expression of Nctc1 at the Igf2/H19/Nctc1 locus. Nucleic Acids Res 2013;41:817-826.
- Eun B, Sampley ML, Van Winkle MT, Good AL, Kachman MM, Pfeifer K. The Igf2/H19 muscle enhancer is an active transcriptional complex. Nucleic Acids Res 2013;41:8126-8134.
- Leikina E, Melikov K, Sanyal S, Verma SK, Eun B, Gebert CM, Pfeifer K, Lizunov V, Kozlov M, Chernomordik LV. Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J Cell Biol 2013;200:109-123.
- Xia J, Varudkar N, Baker C, Abukenda I, Martinez C, Natarajan A, Grinberg A, Pfeifer K, Ebert SN. Targeting of the enhanced green fluorescent protein reporter to adrenergic cells in mice. Mol Biotechnol 2013;54:350-360.
Collaborators
- Leonid V. Chernomordik, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- Steve Ebert, PhD, Burnett College of Biomedical Sciences, University of Central Florida, Orlando, FL
- Bjorn Knollmann, MD, PhD, Vanderbilt University Medical Center, Nashville, TN
- Madhulika Srivastava, PhD, National Institute of Immunology, New Delhi, India
Contact
For more information, email kpfeifer@helix.nih.gov or visit pfeiferlab.nichd.nih.gov.