

You are here: Home > Section on Chromatin and Gene Expression
Chromatin Remodeling and Gene Activation

- David J. Clark, PhD, Head, Section on Chromatin and Gene Expression
- Peter Eriksson, PhD, Staff Scientist
- Tara L. Burke, PhD, Postdoctoral Fellow
- Hope A. Cole, PhD, Postdoctoral Fellow
- Razvan V. Chereji, PhD, Visiting Fellow
- Josefina Ocampo, PhD, Visiting Fellow
Our main objective is to understand how genes are activated for transcription in the context of chromatin structure. Chromatin is not only a packaging system for DNA in eukaryotic cells but also participates in gene regulation. Many single-gene studies, including our own, have led to various models describing the events that occur in chromatin when genes are activated, both in yeast and higher organisms, although the extent to which such models can be generalized has been unclear. However, the recent advent of high-throughput technologies has led to a plethora of studies addressing the genome-wide locations of sequence-specific DNA–binding proteins, chromatin-remodeling complexes, and nucleosomes. We are exploiting the new technology to map nucleosomes genome-wide in budding yeast and to determine what happens to nucleosomes on genes when they are activated. We find that many nucleosomes are apparently displaced and that those that remain are re-positioned. We are extending these studies to the roles of transcription activators, ATP-dependent nucleosome remodeling complexes, histone chaperones, and histone modifications in determining the fate of nucleosomes when genes are transcribed. Currently, we are focusing on the roles of the SWI/SNF class of chromatin-remodeling machines, which are mutated in various cancers. Our aim is to describe the molecular mechanisms involved in chromatin disruption when genes are activated.
Our second objective is to continue our study of the histone genes in yeast, as a model for the regulation of cell cycle–dependent genes. The histone genes are subject to both positive and negative regulation. We studied the positive regulatory system in depth, demonstrating that Spt10 and SBF are the key positive regulators of the histone genes during the cell cycle. We are now testing a model for histone gene regulation that we proposed recently, which posits a critical role for histone chaperones as part of a negative feedback control system. We are also studying the changes in chromatin structure that occur during the cell cycle, genome-wide, with particular emphasis on histone gene loci. Thus, we aim to understand the dynamics of chromatin structure during the cell cycle and the roles of trans-activators in this process.
Roles of the SWI-SNF and RSC chromatin-remodeling complexes in nucleosome phasing
RSC and SWI/SNF are related ATP–dependent chromatin-remodeling machines that move nucleosomes, regulating access to DNA. These complexes are conserved from yeast to man, where they play critical roles in gene regulation and other essential processes. Furthermore, it has become clear that mutations in SWI/SNF and related complexes are strongly associated with various cancers, particularly pediatric cancers. In this project, we are addressing the roles of SWI/SNF and RSC in nucleosome phasing relative to transcription start sites in budding yeast, in which it is relatively easy to eliminate the complexes' activities, either using a null mutation for SWI/SNF (snf2) or by depleting cells of an essential subunit (the Rsc8 subunit of RSC). In vivo, gene promoters are typically depleted of nucleosomes (nucleosome-depleted regions or "NDRs"). Thus, nucleosomes on genes are organized into regularly spaced arrays, where the first (+1) nucleosome is located either directly over the transcription start site (TSS) or very close to it. The ordering of nucleosomes with respect to the TSS is referred to as nucleosome phasing. We obtained genome-wide nucleosome maps from wild-type and mutant cells by isolating DNA protected from micrococcal nuclease (MNase) digestion and subjecting it to paired-end sequencing (1).
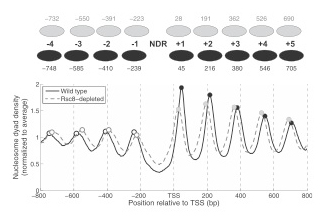
Figure 1. Nucleosome phasing relative to the transcription start site (TSS) shows global shifts of both upstream and downstream nucleosomal arrays toward the nucleosome-depleted region in RSC–depleted cells, with no change in nucleosome spacing.
Average nucleosome dyad density near the TSS in wild-type (solid line) and in RSC–depleted budding yeast cells (dashed line). NDR, nucleosome-depleted region at the gene promoter. Black ovals: nucleosome positions in wild-type cells; grey ovals: nucleosome positions in RSC–depleted cells. Note that the NDR is narrower after RSC depletion and that nucleosome spacing is unaffected (from reference 1).
Rather surprisingly, SWI/SNF has no effect on nucleosome phasing at the global level. To explain the observation, we propose that SWI/SNF regulates relatively few genes. In contrast, RSC depletion results in global nucleosome repositioning; both upstream and downstream nucleosomal arrays shift toward the NDR, with no change in spacing, resulting in a narrower and partly filled NDR (Figure 1). The global picture of RSC–depleted chromatin represents the average of a range of chromatin structures, with most genes showing a shift of the +1 or the −1 nucleosome into the NDR. Using RSC ChIP data reported by others, we showed that RSC occupancy is highest on the coding regions of heavily transcribed genes, though not at their NDRs. We propose that RSC has a role in restoring chromatin structure after transcription. Analysis of gene pairs in different orientations demonstrates that phasing patterns reflect competition between phasing signals emanating from neighboring NDRs. The signals may be in phase, resulting in constructive interference and a regular array, or out of phase, resulting in destructive interference and fuzzy positioning (Figure 2). We propose a modified barrier model, in which a stable complex located at the NDR acts as a bidirectional phasing barrier. In RSC–depleted cells, the barrier has a smaller footprint, resulting in narrower NDRs. Thus, RSC plays a critical role in organizing yeast chromatin.
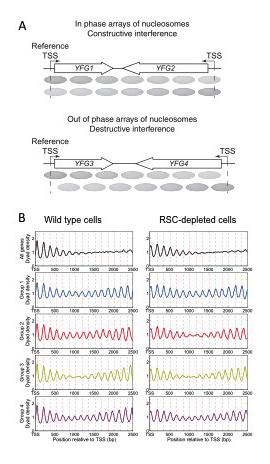
Figure 2. Nucleosome phase interference analysis of convergent gene pairs in wild-type and RSC–depleted cells
A. Phasing of nucleosomes relative to the transcription start site (TSS) occurs from barrier complexes located in the promoters of both genes. The two arrays of nucleosomes may be in phase or out of phase, depending on the distance between the TSSs, resulting in constructive or destructive interference patterns. B. Top panels: average nucleosome center distributions for all convergent gene pairs relative to the TSS. Note that phasing on all genes gradually weakens with distance from the reference TSS at left. The lower four panels correspond to the same genes in the top panels divided into four phase groups according to their phase differences: 0 bp (in phase), 40 bp, 80 bp (almost exactly out of phase) and 120 bp (see reference 1). Note that peaks are now apparent far downstream of the reference TSS at left. The dashed lines indicate the positions of nucleosomes in phase with the reference TSS.
Currently, we are extending these studies to examine the roles of additional remodelers (ISW1, ISW2, and CHD1) in organizing yeast chromatin and to determine whether they have specific roles or are functionally redundant.
Interaction between the large subunit of RNA polymerase II and core histones during transcription through the nucleosome
The question of how RNA polymerase II transcribes through a nucleosome has been studied for many years: the nucleosome is very compact, yet RNA polymerase II can transcribe through it, displacing only a histone H2A-H2B dimer. We investigated how the bulky polymerase achieves this feat. We collaborated with the Studitsky Lab to compare the kinetics of transit through a single nucleosome by purified wild-type and mutated RNA polymerase II (2). The mutations are located in a negatively charged region of the polymerase identified by modeling, which was postulated to interact with the positively charged histones during transit, such that the histones are more likely to be retained after transcription. We found that, although the mutated polymerase transcribes at a similar rate to wild type, the histone octamer is more likely to be displaced from the DNA, consistent with a histone-polymerase interaction during transcription.
Spt10 and SBF control the timing of histone H2A/H2B gene activation in budding yeast.
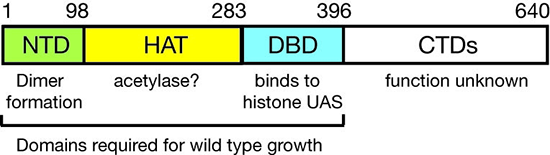
Figure 3. Domain structure of Spt10p
NTD, N-terminal domain; HAT, histone acetylase; DBD, DNA–binding domain; CTDs, C-terminal domains; UAS, upstream activating sequences
We showed that Spt10 is a very unusual trans-activator, in which a histone acetylase (HAT) domain, normally recruited as a co-activator to promoters through an activation domain, is attached directly to a sequence-specific DNA–binding domain (Figure 3). More recently, we addressed the role of Spt10 in the cell cycle–dependent regulation of the histone genes, a regulation that is necessary to provide histones for nucleosome assembly during DNA replication. Histones H2A and H2B are expressed from divergent promoters at the HTA1-HTB1 and HTA2-HTB2 loci. We showed that Spt10 and the cell-cycle regulator SBF (a Swi4-Swi6 heterodimer) have overlapping binding sites in the HTA1-HTB1 promoter. Both SBF and Spt10 are bound in cells arrested with alpha-factor, apparently awaiting a signal to activate transcription. Soon after removal of alpha-factor, SBF initiates a small, early peak of HTA1 and HTB1 transcription, which is followed by a much larger peak attributable to Spt10. Both activators dissociate from the HTA1-HTB1 promoter after expression has been activated. Thus, SBF and Spt10 cooperate to control the timing of HTA1-HTB1 expression.
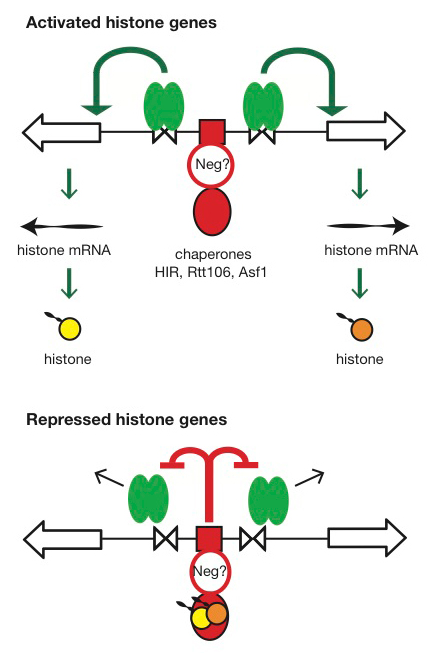
Our current work has two goals: (i) understanding the dynamics of chromatin structure during the cell cycle at the histone genes and genome-wide; (ii) testing our proposed model for histone gene regulation (Eriksson PR et al., Genetics 2012;191:7-20), which posits that the extent to which histone chaperones are saturated with histones is the critical signal for negative feedback control of transcription (Figure 4).
Click image to enlarge.
Figure 4. Hypothesis for the regulation of the HTA1-HTB1 locus
Hypothesis: Negative feedback by histones links the positive and negative regulatory (NEG) systems. The histone genes are activated by Spt10 and SBF bound to the histone UAS elements (open triangles), followed by transcription and translation to produce histones. The NEG system inhibits activation through a putative sequence-specific NEG factor (open red circle) bound to the NEG region (red box), which recruits various histone chaperones (HIR, Rtt106, and Asf1; red oval). Inhibition occurs only if histones are bound to the chaperones. The chaperones are fully charged with histone and maximally inhibitory when there is no replicated DNA available in the cell for nucleosome assembly—when replication is complete (outside S-phase) or if replication forks are stalled (e.g., in the presence of hydroxyurea). Outside S-phase, the activators are displaced or degraded (arrows) (Eriksson PR, Ganguli D, Nagarajavel V, Clark DJ. Regulation of histone gene expression in budding yeast. Genetics 2012;191:7-20).
Publications
- Ganguli D, Chereji RV, Iben JR, Cole HA, Clark DJ. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res 2014;24:1637-1649.
- Chang HW, Kulaeva OI, Shaytan AK, Kibanov M, Kuznedelov K, Severinov KV, Kirpichnikov MP, Clark DJ, Studitsky VM. Analysis of the mechanism of nucleosome survival during transcription. Nucleic Acids Res 2014;42:1619-1627.
Collaborators
- James Iben, PhD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
- Vasily M. Studitsky, PhD, Fox Chase Cancer Center, Temple University Health System, Philadelphia, PA
Contact
For more information, email clarkda@mail.nih.gov or visit ucge.nichd.nih.gov.