

You are here: Home > Section on Molecular Genetics of Immunity
Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Natarajan Ayithan, PhD, Visiting Fellow
- Mahesh Bachu, PhD, Visiting Fellow
- Monica Gupta, PhD, Visiting Fellow
- Rui Kamada, PhD, Visiting Fellow
- Naoyuki Sarai, PhD, Visiting Fellow
- Yuko Yoshida, PhD, Visiting Fellow
- Hiroaki Yoshii, PhD, Visiting Fellow
- Tamaki Ito, PhD, Japan Society for the Promotion of Science–NIH Fellow
- Ankur Narain, BS, Postbaccalaureate Fellow
Macrophages (MΦ) and dendritic cells (DC) respond to pathogen stimuli by producing cytokines, including interferons (IFNs), IL-1, IL-6, and TNF-alpha, to impart anti-viral and anti-microbial status to the host. Our goal is to study the molecular pathways that direct the development and function of MΦ and DC. Our long-term interest has been the role of the transcription factor IRF8 in innate immunity. IRF8 is expressed in MΦ and DC at high levels and is required for the production of both type I and type II IFNs. IRF8 is essential for mounting the first line of defense against various invading pathogens prior to the initiation of antigen-specific immune responses. Autophagy is one of mechanisms by which MΦ and DC eliminate pathogens. It is thought that autophagy plays a role in host resistance against HIV-1 infection. We are interested in the role of IRF8 in autophagy and innate immune responses.
Transcriptionally active genes are embedded in chromatin that is dynamically exchanged, whereas silenced genes are surrounded by more stable chromatin. The chromatin environment influences transcriptional processes and controls epigenetic regulation. We are interested in BRD4, a chromatin-binding protein associated with transcribed genes. We are also interested in histone H3.3, the variant histone that is selectively associated with actively expressed genes. BRD4 is a 200 kDa nuclear protein carrying two tandem bromodomains through which it binds to acetylated chromatin. BRD4 also interacts with the transcription elongation factor P-TEFb and regulates transcription of many genes, including those induced by external stimuli. BRD4 is implicated in transcriptional memory across cell division because it stays on condensed chromosomes during mitosis and affects gene expression in daughter cells. Despite close structural similarity with the standard H3.1 and H3.2 histones, histone H3.3 has an extraordinary property in that it is incorporated into nucleosomes and DNA only in actively transcribed genes. In contrast, H3.1, H3.2, and other standard core histones are incorporated into nucleosomes during DNA replication. The difference between H3.3 and H3.1/2 reflects distinct chromatin activities during replication and transcription. Although there is mounting recognition of the importance of transcription-coupled histone incorporation, the process and its physiological significance are still shrouded in mystery. Our goal is to elucidate the activity of BRD4 and histone H3.3 in the context of transcriptional activation and epigenetic memory.
BRD4 supports elongation of the eRNA and the coding RNA
BRD4 has two bromodomains through which it binds to the acetylated histones H3 and H4. BRD4 also interacts with P-TEFb, the elongation factor composed of Cyclin T and CDK9. The interaction relieves the 5′ paused RNA polymerase II (Pol II) to trigger elongation. Recent studies from various laboratories showed that BRD4 promotes cancer cell growth, including several forms of leukemia, primarily by promoting expression of the oncogene c-MYC. Moreover, recently developed bromodomain small-molecule inhibitors have been found to be powerful anti-leukemia drugs in animal models. To further study the extent of BRD4 involvement in elongation, we performed RNA-seq and ChIP-seq analyses of chromatin-bound RNA in quiescent and serum-stimulated fibroblasts. We were particularly interested in the enhancer regions that carry epigenetic histone marks such as H3K4me1 and H3K27ac and affect expression of the coding genes. Our results showed that serum stimulation activates transcription of many cell growth–regulated genes in their coding region as well as their enhancer (e) sequences either in the upstream or downstream. Transcription of both coding and enhancer (eRNA) RNAs were dependent on BRD4’s interaction with acetylated histones, given that JQ1, a small-molecule inhibitor of BRD4, blocked both RNAs. In addition, ChIP-seq data showed that BRD4 localized to the coding and enhancer regions that overlap with the localization of Pol II, supporting the view that BRD4 takes part in the elongation processes by virtue of its ability to bind to acetylated histones. To confirm the broad role of BRD4 in RNA transcription, nascent RNA was labeled with Br-UTP and immunoprecipitated by BRD4 antibody and then with BrdU antibody. Double immunoprecipitated RNA samples were analyzed by deep sequencing. We found that BRD4 is bound to newly synthesized eRNAs as well as to coding RNAs. The data led us to a model wherein BRD4 directs elongation of eRNA and coding RNA that is guided by BRD4's biding to acetylated histones, thereby contributing to the transcription of epigenetically regulated genes.
Histone methyltransferase WHSC1 interacts with the chromatin assembly factor HIRA to drive H3.3 deposition in interferon-stimulated genes.
H3.3 is highly similar to the canonical histone H3 (H3.1 and H3.2) in its structure, differing only in a few amino acids. Unlike the canonical histone H3, however, H3.3 (encoded by two genes in the human and mouse) is synthesized outside the S phase and is incorporated into chromatin along with transcription. It is thought that H3.3 associates with the preexisting canonical histone H4 to form a new nucleosome, presumably along with the H2 variant H2AZ and the preexisting H2B. H3.3 incorporation into chromatin requires specific chromatin assembly factors distinct from those involved in replication-coupled histone deposition. HIRA is a major H3.3–specific histone chaperon dedicated to transcription-coupled H3.3 incorporation. HIRA carries the WD40 domain and B domain, through which it interacts with three other subunits to act as a chromatin assembly factor. Two other H3.3–specific chaperons, ATRX and Daxx, are reported to be involved in H3.3 incorporation into the telomeres and pericentric heterochromatin.
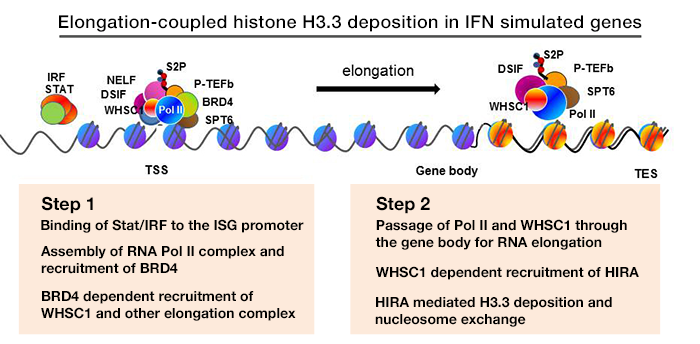
Click image to enlarge.
Figure 1. Elongation-coupled histone H3.3 deposition in IFN–stimulated genes
We studied transcription-coupled H3.3 incorporation, using the mode and kinetics of deposition in IFN–stimulated genes (ISGs), such as Ifit1, Mx1, Oas1, and Stat1, as a model. We showed that H3.3 is incorporated into all four ISGs as soon as their transcription begins. The notable features of IFN–induced H3.3 deposition are that (i) it occurs with a positional gradient, such that the deposition is the greatest in the 3′ transcription end site with corresponding reduction towards the transcription start side (TSS), and (ii) H3.3 deposition continues long after transcription ceases, even 48 h after IFN stimulation. Essentially the identical pattern of H3.3 deposition was seen for the c-Fos and c-Jun genes after UV stimulation. We noted that trimethylation of H3K36 is induced after stimulation and that the temporal and positional patterns of increase closely correlate with H3.3 incorporation. Methylation of H3K36 is catalyzed by the SET2 family of histone methylases, including WHSC1 (also known as NSD2). We found that Whsc1−/− mouse embryonic fibroblasts (MEFs) failed to deposit H3.3 after IFN stimulation, while Whsc1+/+ MEFs supported the expected H3.3 deposition. Further, WHSC1 interacted with the chromatin-binding factor BRD4 and is thus critical for the recruitment of the transcription-elongation factor P-TEFb. In the absence of WHSC1, ISG elongation was strongly impaired, resulting in a severe loss of ISG mRNA induction. Chromatin-immunoprecipitation (ChIP) analysis demonstrated that WHSC1 was recruited to the ISGs upon IFN stimulation. WHSC1 was distributed throughout the coding regions and remained on the ISGs long after the cessation of ISG transcription. In parallel with WHSC1 recruitment, HIRA bound to the ISGs after IFN stimulation and remained on ISGs as long as WHSC1 did. Furthermore, WHSC1 directly interacted with HIRA through the internal PHD domain and the HMG domain in a manner independent of the interaction with BRD4. Consistent with the interaction of WHSC1 and HIRA, WHSC1 and HIRA bound to the ISGs as a protein complex, as confirmed by re-ChIP assays. Together, our studies reveal two phases of H3.3 deposition: (i) the initial elongation-coupled phase of deposition, and (ii) a subsequent switch to the post-transcription phase of deposition. It is clear that WHSC1 plays a central role in both phases. Our aim is to extend our analysis from tissue culture models to in vivo animal models to address the modes and the mechanisms of H3.3 deposition. To this end, we constructed Hiraf/f mice that allow conditional disruption of the gene. With the mice, we plan to study HIRA–dependent H3.3 deposition in various immune cells.
Construction of H3.3–HA knock-in mice and validation of the experimental model
To study the activity of H3.3 in vivo, we generated mouse strains in which the endogenous H3.3 genes (H3.3A, H3f3a and H3.3B, H3f3b) are replaced by hemagglutinin (HA)-tagged H3.3 genes. The knock-in targeting vectors contain HA–tagged H3.3 cDNA and an IRES–driven GFP to allow independent monitoring of the expression of the inserted genes. The inserted H3.3A–HA and H3.3B–HA fragments are flanked by the LoxP sites to allow conditional knockout of the genes. The original targeting vectors had the neomycin- and hygromycin-resistant genes for ES cell selection but were removed by FRT–based recombination to avoid potential adverse effects in the mouse. Heterozygous mice for the H3.3A–HA and H3.3B–HA loci are viable and fertile, although the birth rate of homozygous mice appears significantly lower than that expected for Mendelian inheritance. We confirmed that H3.3–HA is expressed at variable levels throughout adult tissues. Preliminary ChIP analysis performed with anti–HA antibody indicated that H3.3–HA is deposited in induced genes in bone marrow–derived MΦ stimulated with IFNg and toll-like receptor (TLR) signaling. We established MEFs that carry homozygous and heterozygous H3.3A–HA and H3.3B–HA, and we plan to analyze the MΦ and MEFs for global H3.3 deposition, using the ChIP-seq approach.
The role of IRF8 and in protection against infectious pathogens
IRF8, a transcription factor of the IRF family, is highly expressed in MΦ and DC and is shown to be important for host resistance against various pathogens, including Mycobacterium tuberculosis, Salmonella, and Listeria, as well as against RNA and DNA viruses. Given that Ebola viruses (EBOV) infect MΦ and DC as the initial site of infection and inhibit IFN induction and IFN signaling, we became interested in the virus, also because it causes severe hemorrhagic fever with high fatality. EBOV virus–like particles (VLPs), which are genome-free viral proteins composed of the matrix protein (VP40), membrane glycoprotein, and nucleoprotein (NP), serve as a promising vaccine candidate against EBOV infection. When given prior to EBOV infection, VLPs fully protect rodents and nonhuman primates against the EBOV disease. Recent studies show that, even when given following lethal EBOV infection, EBOV can protect mice. We investigated the mechanisms by which VLPs provide post-exposure protection, focusing on the role of IFN and IRF8. We found that VLP administration fails to protect Ifnar−/− mice, which lack type I interferon (IFN) signaling. In wild-type mice, VLPs accelerated induction of many IFN–stimulated genes (ISGs) in liver and spleen. The ISGs included classical anti-viral proteins and negative-feedback factors known to restrict excessive inflammatory responses. VLPs did not induce ISGs in Ifnar−/− mice, and consequently uncontrolled viral growth and elevated proinflammatory cytokines and chemokines ensued in the mice, indicating that type I IFNs broadly attenuate inflammatory responses through IFN–induced negative-feedback factors. We also found that VLPs were unable to establish preventive anti–EBOV immunity in Irf8−/− mice, illustrating the role of IRF8 in initial IFN activation in critical cell types. Supporting the in vivo results, we showed that VLPs activate type I IFN genes and ISGs through the toll-like receptor signaling in MΦ and DC. Together, VLPs afford post-exposure protection by expeditious induction of type I IFNs in the host.
IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation: the role in multiple sclerosis
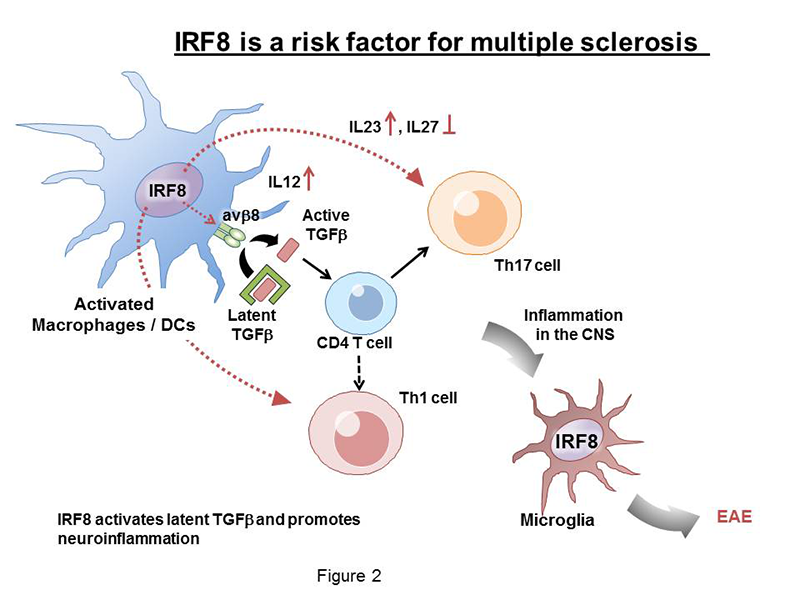
Click image to enlarge.
Figure 2. IRF8 as a risk factor for multiple sclerosis
Multiple sclerosis (MS) is a debilitating chronic disease that adversely affects not only mobility but also mental conditions. The disease often starts in young adults and progressively worsens over years and decades. MS is more prevalent in women than in men. Although various treatments are available that can ameliorate the symptoms, there is no cure for the disease. MS causes demyelination of neurons and their eventual destruction, which is believed to be attributable to autoimmune responses. Clinical and animal model studies have shown that activation of Th17 T helper cells and infiltration of T cells and activated myeloid cells are part of complex mechanisms that drive MS disease progression. Other lines of evidence indicated that cytokines, particularly, TGF-β, IL-17, and IL-23, play important roles in the initiation and progression of MS. Our study was prompted by several recent epidemiological reports that identified IRF8 as one of new risk factors for the MS. We thus investigated the role of IRF8 in experimental allergic encephalomyelitis (EAE), a mouse model of MS. In the animal model, a single injection of the myelin antigen with adjuvants causes neuroinflammatory conditions that resemble MS, demonstrating limb paralysis associated with demyelination and inflammation of the central nervous system (CNS). We found that Irf8 knockout mice are completely resistant to EAE, indicating that IRF8 plays an active role in EAE. Analysis of Irf8 conditional knockout mice found that IRF8 in MΦ and dendritic cells are responsible for EAE, rather than IRF8 expressed in T helper cells. Our data showed that IRF8 is critical for the activation of TGF-β, required for the initial sensitization of Th17 and other T helper cells. This is because IRF8 was required for the expression of the integrin avb8, which can convert the latent TGF-β into an active TGF-β. The integrin, expressed on the surface of MΦ and dendritic cells, releases active TGF-β in the vicinity of T cells and activates TGF-β signaling directly in naive T cells to initiate the disease. Accordingly, Irf8 knockout mice failed to activate TGF-β signaling in T cells. By stimulating integrin expression in MΦ and dendritic cells, IRF8 plays a decisive role in the initiation of EAE.
Additional studies found that IRF8 also drives disease progression in the subsequent stages. First, IRF8 promoted production of the proinflammatory cytokine IL-23 and inhibited anti-inflammatory cytokine IL-27, which are reported to regulate development and maintenance of Th17 T cells. Second, IRF8 was expressed in microglia, immune cells in the brain that are activated in various CNS diseases. We found that microglia in Irf8 knockout mice were not activated during EAE, consistent with earlier reports that IRF8 is required for microglia activation. Thus, IRF8 promotes initiation and progression of EAE at multiple steps through distinct mechanisms (see Diagram in Figure 2). Taken together, the studies provide mechanistic insight into how IRF8 acts as a risk factor for MS.
Publications
- Ayithan N, Bradfute SB, Anthony SM, Stuthman KS, Dye JM, Bavari S, Bray M, Ozato. K. Ebola virus-like particles stimulate type I interferons and proinflammatory cytokine expression through the toll-like receptor and interferon signaling pathways. J Interferon Cytokine Res 2014;34(2):79-89.
- Sarai N, Nimura. K, Tamura T, Kanno T, Patel MC, Heightman TD, Ura K, Ozato, K. WHSC1 links transcription elongation to HIRA mediated histone H3.3 deposition. EMBO J 2013;32:2392-2406.
- Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey A, Xiong H, Munasinghe J, Yazawa I, O'Donovan MJ, Maximova OA, Sharma S, Zhu J, Wang H, Morse HC 3rd, Ozato K. The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation. Immunity 2013;40(2):187-198.
- Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, Sasaki H, Matsunaga S, Yoshinari M, Ryo A, Nakazawa M, Ozato K, Tamura T. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun 2014;5:4978.
- Alsarraj J, Faraji F, Geiger TR, Mattaini KR, Williams M, Wu J, Ha NH, Merlino T, Walker RC, Bosley AD, Xiao Z, Andresson T, Esposito D, Smithers N, Lugo D, Prinjha R, Day A, Crawford NP, Ozato K, Gardner K, Hunter KW. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the LINC complex at the inner face of the nuclear membrane. PLoS One 2013;8(11):e80746.
Collaborators
- Steven B. Bradfute, PhD, United States Army Medical Institute of Infectious Diseases, Fort Detrick, MD
- Herbert Morse II, MD, Laboratory of Immunopathology, NIAID, Rockville, MD
- Dinah S. Singer, PhD, Experimental Immunology Branch, NCI, Bethesda, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
Contact
For more information, email ozatok@mail.nih.gov or visit ozatolab.nichd.nih.gov.