

You are here: Home > Section on Formation of RNA
Physiological, Biochemical, and Molecular-Genetic Events Governing the Recognition and Resolution of RNA/DNA Hybrids

- Robert J. Crouch, PhD, Head, Section on Formation of RNA
- Susana M. Cerritelli, PhD, Staff Scientist
- Naushaba Hasin, PhD, Postdoctoral Fellow
- Ryo Uehara, PhD, Postdoctoral Fellow
- Kiran Sakhuja, MS, MSc, Biologist
Damaged DNA is a leading cause of many human diseases and disorders. We study the formation and resolution of RNA/DNA hybrids, which occur during DNA and RNA synthesis. Such hybrid molecules may lead to increased DNA damage but may also play critical roles in normal cellular processes. We are interested in how RNA/DNA hybrids are resolved and in the role that ribonucleases H (RNases H) play in their elimination (Cerritelli and Crouch, FEBS J 2009;276:1494). Two classes of RNases H are present in most organisms. Our studies showed that mice deleted for the Rnaseh1 gene arrest embryonic development at day 8.5 because they fail to amplify mitochondrial DNA. Others have found that the Aicardi-Goutières Syndrome (AGS), a severe neurological disorder with symptoms appearing at or soon after birth, can be caused by defective human RNase H2. To understand the mechanisms and functions of RNases H, we employ molecular-genetic and biochemical tools and yeast and mouse models in our research.
There are several types of RNA/DNA hybrid structures, which are formed and processed differently. (i) Simple RNA/DNA hybrids consist of one strand of RNA paired with one strand of DNA. The HIV-AIDS reverse transcriptase (RT) forms these hybrids when copying its genomic RNA into DNA. The RT also has an RNase H domain that is structurally and functionally similar to the class I cellular RNase H and is necessary for several steps of viral DNA synthesis. (ii) R-loop hybrids have two DNA strands separated, with one hybridized to RNA while the other is in single-stranded form. The structures sometimes form during transcription and can lead to chromosomal breakage. However, they are also part of the normal recombination process of switching (recombination) from one form of immunoglobulin to another, resulting in different isoforms of antibodies. (iii) During replication, single or multiple ribonucleotides become incorporated into DNA (1). The first two types of hybrid are substrates for class I and II RNases H. The third is uniquely recognized by type 2 RNases H.
Contrasts between Class I and Class II RNases H
Our previous work on RNase H1 showed that the enzyme recognizes the 2′-OH of four consecutive ribonucleotides while the DNA strand is distorted to fit in a pocket of the enzyme. Thus, the enzyme requires more than one ribonucleotide for cleavage of RNA in RNA/DNA hybrids. RNases H1 consist of a single polypeptide in both eukaryotes and prokaryotes. In contrast, RNase H2 is a complex of three different polypeptides in eukaryotes but is a single polypeptide in prokaryotes. The catalytic subunit of the heterotrimeric RNase H2 of eukaryotes is similar in its primary amino acid sequence to the prokaryotic enzyme. RNase H2 can recognize and cleave a single ribonucleotide or the transition from the ribonucleotide in the case of RNA-primed DNA synthesis (e.g., rrrrrDDDD in DNA—italics indicate transition from ribonucleotide to deoxyribonucleotide).
Dual activities of RNase H2 and Aicardi-Goutières syndrome
Eukaryotic RNases H2 recognize and resolve RNA hybridized with or covalently attached to DNA—two chemically distinct structures—using the same catalytic mechanism of hydrolysis. RNase H2 mutations that reduce catalytic activity or fail to properly interact with in vivo substrates cause Aicardi-Goutières syndrome (AGS). Of the 29 different RNase H2 mutations associated with AGS, eight are in the A subunit, 14 in the B subunit, and 7 in the small C subunit (2). We previously expressed in E. coli and purified human RNases H2 with mutations corresponding to several of those seen in AGS patients. We found only one with significant loss of RNase H2 activity (3). Using the structure we determined, we located all known mutations in RNase H2 causing AGS (2). The wide distribution of these mutations suggests that modest changes in stability, interaction with other unknown proteins, and loss of catalysis can all cause AGS.
We are developing mouse models of AGS to clarify which defects are associated with each RNase H2 activity. Mice bearing the Rnaseh2A G37S mutation in homozygous form are either stillborn or die within a few hours of birth. Mutations in another gene, Trex1, also cause AGS, and it has been shown that homozygous knockout (KO) mice are viable but die after a few weeks owing to a cardiomyopathy that can be prevented by blocking either innate or adaptive immune responses. In contrast, Rnaseh2 KO mice die during embryogenesis; this and the stillborn phenotype of the G37S mutation suggest a more severe defect than that seen in Trex1-KO mice. We attempted to rescue the stillborn phenotype by eliminating innate or adaptive immune responses. The mice are no different from the innate- or adaptive-competent mice, suggesting the possibility of a different issue between the Trex1– and Rnaseh2–mutant mice. However, several interferon-stimulated genes (ISGs) are increased in mouse embryonic fibroblasts (MEFs) derived from G47S homozygous embryos, supporting a role for innate immunity in the AGS phenotype. We are continuing to examine these mice for specific defects in various tissue and organs associated with the mutation.
To distinguish the defects that, in vivo, persistent RNA/DNA hybrids cause from those caused by single ribonucleotides joined to DNA, we modified RNase H2 to make an enzyme that could only cleave one type of substrate. Based on a rational design comparing the structures of RNases H2 and H3, we unlinked the two activities to yield an enzyme that processes RNA/DNA, but leaves single ribonucleoside monophosphates (rNMPs) attached to DNA. RNases H2 and H3 have similar 3D structures, but RNase H3 does not cleave single rNMPs in DNA. We first examined the in vitro activities of our new RNase H2 mutant using, among others, the ribonucleotide excision repair (RER) assay recently developed by our collaborators Peter Burgers and Justin Sparks (4). The in vitro results show complete lack of removal of single ribonucleotides but only modest reduction in hydrolysis of RNA/DNA hybrids. In vivo, our RNase H2 mutant gave the signature 2–5 bp deletions in the yeast can1 gene associated with incorporation of ribonucleotides into DNA in the absence of RNase H2 (5). The mutant enzyme resolves RNA/DNA hybrids, which are substrates of both RNase H1 and RNase H2. However, our mutant RNase H2 identified a unique set of hybrids that formed when homologous recombination is defective resulting from loss of Sgs1 helicase; the hybrids can only be processed by RNase H2, most likely because it has special access via contacts with other cell components. Thus, the synthetic defect observed in sgs1Δ:rnh201Δ strains results from problems associated with persistent R-loops. Given that our RNase H2–mutant enzyme resolves the R-loops, the synthetic defect is absent when the mutant RNase H2 is present in a strain deleted forsgs1.
The RNASEH2A G37S mutation, which is found in a few AGS patients, results in limited activity of the encoded enzyme in vitro (3). We investigated how the corresponding mutation in yeast RNase H2 (G42S) protein functions. In vitro, it displayed very poor activity on RNA/DNA hybrids and on cleavage of single rNMPs in DNA. In vivo, it was almost as ineffective as a deletion of an RNase H2 subunit for removing single rNMPs (measured by 2–5 bp deletions in the CAN1 gene), had limited activity in removing R-loops accessible to either RNase H2 or H1, yet displayed remarkably good activity when it interacted with other proteins involved in R-loops associated with DNA replication/repair.
Very recently, we developed a mouse line that is unable to initiate removal of rNMPs in DNA and are examining the effects of this mutation on viability as well as other possible defects.
Mitochondrial DNA and RNase H1 in mouse
During mouse embryonic development, RNase H1 is required for progression beyond day E8.5. A single transcript of the mouse Rnaseh1 gene is translated to make two nearly identical proteins, one of which is localized to the nucleus and the other to the mitochondrion. We previously showed that the Rnaseh1–deleted embryos fail to amplify mtDNA (mitochondrial DNA), causing developmental arrest. Nuclear DNA replicated normally in Rnaseh1 knockout embryos. We are examining loss of the Rnaseh1 gene during B-cell development to follow the process of RNase H1 depletion in a simpler system. Following conditional deletion of Rnaseh1 at an early stage of B-cell development in mouse B-cells, we found that resting, naive B-cells are formed but that they are unable to become activated to class switch to other isotypes (e.g., IgG), and sera from these mice have a major deficit in antibodies. We are currently determining whether the loss of mitochondrial DNA is the explanation for the inability of the resting B cells to be completely activated.
In collaboration with Ian Holt, we are examining the roles of RNase H1 in mitochondrial DNA replication by using, among other techniques, analysis of intermediates on two-dimensional gels. Our findings thus far indicate that elevated expression of RNase H1 in mitochondria alters mtDNA replication. In addition, data obtained in Holt's laboratory supporting the existence of RNA/DNA hybrids as replication intermediates indicate that RNase H1 is important for removal of RNA primers of mtDNA replication.
RNase H1 and RecBCD synthetic lethality
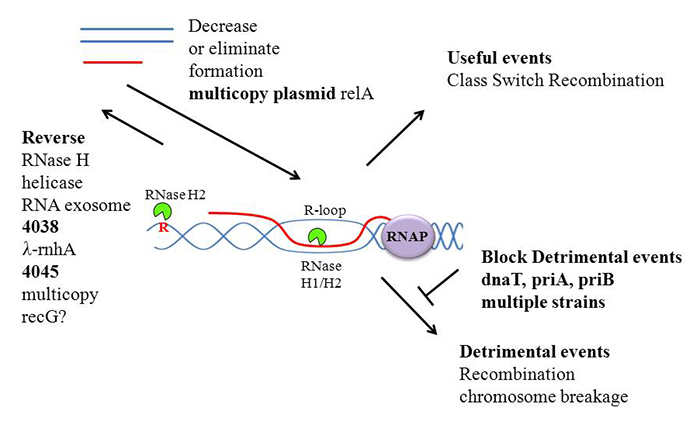
Click image to enlarge.
Figure 1. Formation and prevention of DNA damage created by R-loops
Genes encoding RNase H1 and RecBCD, when inactivated together in E. coli, lead to (synthetic) lethality. Suppressor mutations of the lethality include mutation of each of the three genes (priA, priB, and priC) required for primosome formation. Primosomes are used to initiate restart replication when a replication fork collapses. Because RNase H1 is absent, numerous R-loops form that look like replication collapse products, thereby creating an unusual number of replication restarts. As shown in Figure 1, replication restart must be detrimental to the growth of rnhA-recBCD double mutants, presumably because replication forks collide with one another or with transcription events. R-loops are formed frequently when ribosomal RNA is being synthesized, especially at high growth rates. Increasing the synthesis of ppGpp, a product of the expression of RelA, reduces initiation frequency of transcription of rRNA genes, resulting in fewer R-loops, allowing rnhA-recBCD strains to survive. We are continuing to mine the data we derived by sequence determination of other suppressor strains of E. coli rnhA-recBCD synthetic lethality.
Additional Funding
- Scientific Director's Intramural Award
Publications
- Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes from eukaryotes. FEBS J 2009;276:1494-1505.
- Cerritelli SM, Chon H, Crouch, RJ. A new twist for topoisomerase. Science 2011;332:1510-1511.
- Figiel M, Chon H, Cerritelli SM, Cybulska M, Crouch RJ, Nowotny M. The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi-Goutières syndrome defects. J Biol Chem 2011;286:10540-10550.
- Chon H, Vassilev A, DePamphilis ML, Zhao Y, Zhang J, Burgers PM, Crouch RJ, Cerritelli SM. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res 2009;37:96-110.
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. RNase H2-initiated ribonucleotide excision repair. Mol Cell 2012;47:980-986.
- Chon H, Spark JL, Rychlik M, Nowotny M, Burgers PM, Crouc RJ, Cerritelli SM. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 2013;41:3130-3143.
Collaborators
- Peter Burgers, PhD, Washington University, St. Louis, MO
- Ian Holt, PhD, MRC-National Institute of Medical Research, London, United Kingdom
- Richard J. Maraia, MD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
- Herbert C. Morse, MD, Laboratory of Immunopathology, NIAID, Bethesda, MD
- Justin Sparks, PhD, Washington University, St. Louis, MO
- Roger Woodgate, PhD, Laboratory of Genomic Integrity, NICHD, Bethesda, MD
- Nan Yan, PhD, UT Southwestern Medical Center, Dallas, TX
Contact
For more information, email crouch@helix.nih.gov or visit sfr.nichd.nih.gov.