

You are here: Home > Section on Chromatin and Gene Expression
Chromatin Remodeling and Gene Activation

- David J. Clark, PhD, Head, Section on Chromatin and Gene Expression
- Peter Eriksson, PhD, Research Fellow
- Hope A. Cole, PhD, Postdoctoral Fellow
- Dwaipayan Ganguli, PhD, Visiting Fellow
- Nagarajavel Vivekananthan, PhD, Visiting Fellow
Our main objective is to understand how genes are activated for transcription in the context of chromatin structure. In eukaryotic cells, chromatin is not just a packaging system for DNA but also participates in gene regulation. Many single-gene studies, including our own, have led to various models describing the events that occur in chromatin when genes are activated, both in yeast and higher organisms. But the extent to which such models can be generalized has been unclear. However, the recent advent of high-throughput technologies has led to a plethora of studies addressing the genome-wide locations of sequence-specific DNA binding proteins, chromatin remodeling complexes, and nucleosomes. We are exploiting the new technology to map nucleosomes genome-wide in budding yeast and to determine what happens to nucleosomes on genes when the latter are activated. We find that many nucleosomes are apparently displaced and that those that remain are re-positioned. Currently, we are extending these studies to examine the roles of transcription activators (specifically Gcn4), ATP–dependent nucleosome remodeling machines, histone chaperones, and histone modifications in determining the fate of nucleosomes when genes are transcribed. Our aim is to describe the molecular mechanisms involved in chromatin disruption when genes are activated.
Our second objective is to continue our study of the histone genes in yeast, as a model for the regulation of cell cycle–dependent genes. The histone genes are subject to both positive and negative regulation. We have studied the positive regulatory system in depth, demonstrating that Spt10 and SBF are the key positive regulators of histone genes during the cell cycle. We are now testing our recently proposed model for histone gene regulation, which posits a critical role for histone chaperones as part of a negative feedback control system. We are also studying the changes in chromatin structure that occur during the cell cycle genome-wide, with particular emphasis on the histone gene loci. Thus, we aim to understand the dynamics of chromatin structure during the cell cycle and the roles of trans-activators in this process.
Transcriptional activation and SWI/SNF–dependent nucleosome mobilization
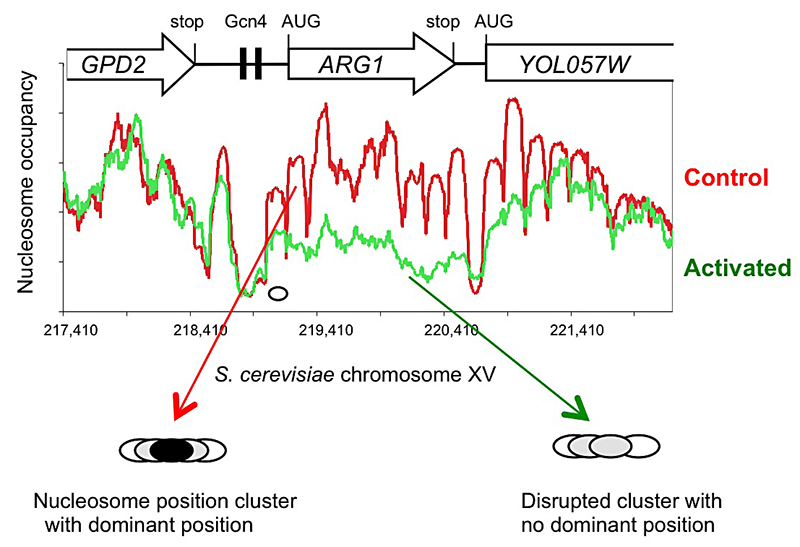
Click image to enlarge.
Figure 1. Activation of the yeast ARG1 gene with 3-aminotriazole results in dramatic loss of nucleosome occupancy and disruption of nucleosome positioning over the coding region, which extends into the downstream YOL057W gene.
An example of genome-wide data obtained using paired-end sequencing (1). Oval: nucleosome drawn to scale.
Previously, we mapped nucleosome positions on two genes in yeast, CUP1 and HIS3, using a high-resolution mapping technique called monomer extension. We found that nucleosome position maps are more complex than anticipated; instead of the unique positions inferred from the classical low-resolution indirect end-labeling technique, we observed clusters of overlapping positions for each nucleosome. A typical cluster has a dominant position with overlapping minor positions, as is generally observed with reconstituted nucleosomes in vitro. In other words, in some cells the nucleosome is in one position while in others it is in a different position. We suggest that this reflects nucleosome dynamics in cells.
To determine how general the nucleosome position cluster organization is, i.e., whether CUP1 and HIS3 are exceptional or typical, we used the new high-throughput sequencing technology to map nucleosomes genome-wide in a relatively simple experiment involving digestion of nuclei with micrococcal nuclease (MNase). We conducted a genome-wide paired-end sequencing study of nucleosomes derived from cells treated with 3-aminotriazole (3AT), which induces Gcn4–dependent genes, and from control cells (1). The questions addressed were: (i) how general the position cluster organization of yeast chromatin is; and (ii) what effect 3AT induction has on the chromatin structure of activated genes.
Analysis of nucleosome positioning on HIS3 confirmed the position cluster organization which we had reported previously for CUP1 and HIS3. Examination of the well studied PHO5 promoter and the TRP1 gene revealed that both loci exhibit a cluster organization, usually with a clear dominant position, rather than the perfectly positioned nucleosomes inferred from indirect end-labeling. We conclude that each nucleosome may adopt one of several alternative positions within a cluster and that this cluster organization is genome-wide. The implication is that different cells within the population have different chromatin structures. Thus, binding sites for regulatory factors might be nucleosomal in some cells and in the linker in other cells. We propose that the position cluster organization of the yeast genome reflects the presence of alternative nucleosome arrays with the same nucleosome spacing, interspersed by nucleosome-depleted regions.
Induction with 3AT results in a major disruption of nucleosome positioning, sometimes with altered nucleosome spacing (2). The most affected genes (50 in all) exhibit a dramatic loss of occupancy over the transcribed region, sometimes extending into neighboring genes (Figure 1). In contrast, nucleosome-depleted promoters are generally unaffected by induction. A small number of genes are repressed by 3AT; these show similar changes in their chromatin structure, but in reverse; loss of nucleosome occupancy is observed in control cells rather than in 3AT–treated cells. Thus, loss of nucleosome occupancy correlates with gene activation.
We also discovered that the specialized nucleosome at the centromere is essentially perfectly positioned, unlike canonical nucleosomes, which occupy one of several alternative overlapping positions (2). We attribute the perfect positioning of the centromeric nucleosome to the presence of sequence-specific factors built into the nucleosome (Figure 2).
Currently, we are exploring the roles of remodeling complexes that are capable of nucleosome mobilization, such as SWI/SNF, in disruption of chromatin structure. We wrote a review of our work, placing it in the context of the chromatin field (3), and described our methodology in detail.
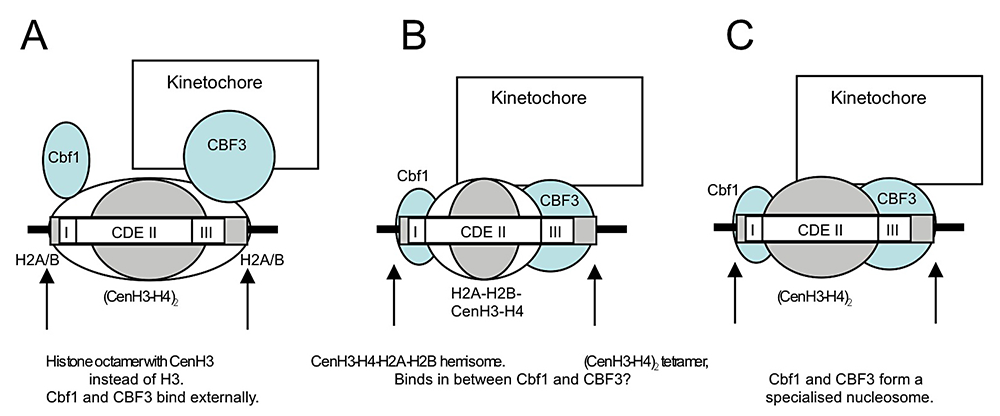
Click image to enlarge.
Figure 2. Models for the perfectly positioned centromeric nucleosome
A: The CenH3 histone octamer model in which CenH3 replaces H3 in an otherwise canonical histone octamer. B: The hemisome model in which a CenH3-H4-H2A-H2B tetramer is bound tightly to sequence-specific factors Cbf1 and CBF3. C: A tetramer containing two molecules of CenH3 and H4 is bound tightly to Cbf1 and CBF3 (2).
Spt10 and SBF control the timing of histone H2A/H2B gene activation in budding yeast.

Click image to enlarge.
Figure 3. Domain structure of Spt10p
NTD, N-terminal domain; HAT, histone acetylase; DBD, DNA-binding domain; CTDs, C-terminal domains; UAS, upstream activating sequences
We showed that Spt10 is a highly unusual trans-activator, in which an HAT domain, normally recruited as a co-activator to promoters through an activation domain, is attached directly to a sequence-specific DNA–binding domain (Figure 3). More recently, we addressed the role of Spt10 in the cell cycle–dependent regulation of the histone genes, which is necessary to provide histones for nucleosome assembly during DNA replication. Histones H2A and H2B are expressed from divergent promoters at the HTA1-HTB1 and HTA2-HTB2 loci. We showed that Spt10 binds to two pairs of UAS elements in the HTA1-HTB1 promoter: UAS1/UAS2 drive HTA1 expression, and UAS3/UAS4 drive HTB1. UAS3 and UAS4 also contain binding sites for the cell-cycle regulator SBF (a Swi4-Swi6 heterodimer), which overlap the Spt10 binding sites. Both SBF and Spt10 are bound in cells arrested with alpha-factor, apparently awaiting a signal to activate transcription. Soon after removal of alpha-factor, SBF initiates a small, early peak of HTA1 and HTB1 transcription, which is followed by a much larger peak due to Spt10. Both activators dissociate from the HTA1-HTB1 promoter after expression has been activated. Thus, SBF and Spt10 cooperate to control the timing of HTA1-HTB1 expression (4).
Our current work has two goals: (i) to understand the dynamics of chromatin structure during the cell cycle at the histone genes and genome-wide; (ii) to test our proposed model for histone gene regulation (4), which posits that the extent to which histone chaperones are saturated with histones is the critical signal for negative feedback control of transcription (Figure 4).

Click image to enlarge.
Figure 4. Hypothesis for the regulation of the HTA1-HTB1 locus
Hypothesis: Negative feedback by histones links the positive and negative regulatory systems. The histone genes are activated by Spt10 and SBF bound to the histone UAS elements (open triangles), followed by transcription and translation to produce histones. The NEG system inhibits activation through a putative sequence-specific NEG factor (open red circle) bound to the NEG region (red box), which recruits various histone chaperones (HIR, Rtt106 and Asf1; red oval). Inhibition occurs only if histones are bound to the chaperones. The chaperones are fully charged with histone and maximally inhibitory when there is no replicated DNA available in the cell for nucleosome assembly—when replication is complete (outside S-phase) or if replication forks are stalled (e.g., in the presence of hydroxyurea). Outside S-phase, the activators are displaced or degraded (arrows) (4).
Structure of the C-terminal domain of the linker histone H1
The structure of the C-terminal domain of histone H1 has been a long-term interest of the lab. In collaboration with Jeffrey Hayes, we showed that this domain undergoes a transition from intrinsic disorder to a folded structure when binding to nucleosomal DNA (5).
Publications
- Cole HA, Howard BH, Clark DJ. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res 2011;39:9521-9535.
- Cole HA, Howard BH, Clark DJ. The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc Natl Acad Sci USA 2011;108:12687-12692.
- Cole HA, Nagarajavel V, Clark DJ. Perfect and imperfect nucleosome positioning in yeast. Biochim Biophys Acta 2012;1819:639-643.
- Eriksson PR, Ganguli D, Nagarajavel V, Clark DJ. Regulation of histone gene expression in budding yeast. Genetics 2012;191:7-20.
- Fang H, Clark DJ, Hayes JJ. DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res 2012;40:1475-1484.
Collaborators
- Jeffrey J. Hayes, PhD, University of Rochester Medical Center, Rochester, NY
- Bruce H. Howard, MD, Program on Genomics of Differentiation, NICHD, Bethesda, MD
Contact
For more information, email clarkda@mail.nih.gov or visit ucge.nichd.nih.gov.