

You are here: Home > Section on Molecular Genetics of Immunity
Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tomohiko Kanno, MD, PhD, Staff Scientist
- Natarajan Ayithan, PhD, Visiting Fellow
- Monica Gupta, PhD, Visiting Fellow
- Rui Kamada, PhD, Visiting Fellow
- Mira Patel, PhD, Visiting Fellow
- Naoyuki Sarai, PhD, Visiting Fellow
- Yuko Yoshida, PhD, Visiting Fellow
- Ankur Narain, BS, Postbaccalaureate Fellow
Macrophages (MФ) and dendritic cells (DC) respond to pathogen stimuli by producing cytokines, including interferons (IFNs), IL-1, IL-6, and TNF-alpha, to impart anti-viral and anti-microbial status to the host. Our goal is to study the molecular pathways that direct the development and function of MФ and DC. Our long-term interest has been the role of a transcription factor IRF8 in innate immunity. IRF8 is expressed in MФ and DC at high levels and is required for production of both type I and type II IFNs. IRF8 is essential for mounting the first line of defense against various invading pathogens prior to the initiation of antigen-specific immune responses. Autophagy is one of mechanisms by which MФ and DC eliminate pathogens. It is thought that autophagy plays a role in host resistance against HIV-1 infection. We are interested in the role of IRF8 in autophagy and innate immune responses.
Transcriptionally active genes are embedded in chromatin that is dynamically exchanged, whereas silenced genes are surrounded by more stable chromatin. The chromatin environment influences transcriptional processes and controls epigenetic regulation. We are interested in BRD4, a chromatin-binding protein associated with transcribed genes. We are also interested in histone H3.3, the variant histone that is selectively associated with actively expressed genes. BRD4 is a 200 kDa nuclear protein carrying two tandem bromodomains through which it binds to acetylated chromatin. BRD4 also interacts with the elongation factor P-TEFb and regulates transcription of many genes, including those induced by external stimuli. BRD4 is implicated in transcriptional memory across cell division because it stays on condensed chromosomes during mitosis and affects gene expression in daughter cells. Despite close structural similarity with the standard H3.1 and H3.2, histone H3.3 has an extraordinary property in that it is incorporated into nucleosomes and DNA only in actively transcribed genes. In contrast, H3.1, H3.2, and other standard core histones are incorporated into nucleosomes during DNA replication. The difference between H3.3 and H3.1/2 reflects distinct chromatin activities during replication and transcription. Although there is mounting recognition of the importance of transcription-coupled histone incorporation, the process and its physiological significance are still shrouded in mystery. Our goal is to elucidate the activity of BRD4 and histone H3.3 in the context of transcriptional activation and epigenetic memory.
IRF8 is a master regulator of autophagy genes required for autophagosome-lysosomal functions.
Active in most cells, autophagy is a conserved catabolic process designed to eliminate misfolded and damaged self proteins. The process is essential during embryonic development—mice with the disrupted autophagy gene ATG5 are embryonic lethal. MΦ and DC employ autophagy as a mechanism of host resistance against invading pathogens. Autophagy is activated by pathogen infection and by IFNg in MΦ, triggering a cascade of events leading to autophagosme formation, fusion with lysosomes, and protein degradation via ubiquitin conjugation and sequestration with SQSTM1 (p62). The processes are regulated by more than 20 genes specializing in executing autophagy. While autophagy helps to clear invading pathogenes, some pathogens utilize autophagy to enhance their growth and survival. Microarray and ChIP-on chip analyses on IRF8+/+ and IRF8−/− dendritic cells revealed that IRF8 regulates expression of some autophagy-related genes. The extension of these analyses led to the observations that IRF8 stimulates at least 17 autophagy genes in MФ. Further tests found that of the 17 genes, 12 could be rescued for expression in IRF8−/− MФ upon IRF8 transfer. IRF8 bound to to the promoter regions of seven of these genes, demonstrating that IRF8 is a major regulator of autophagy gene expression. Accordingly, IRF8−/− MФ are defective in autophagosome formation, the process critical for capturing of misfolded proteins. IRF8−/− MФ are found to be also defective in lysosomal fusion, which is required for final degradation of the captured proteins. In summary, IRF8 is required for full autophagy function in MФ and DC, thereby critically contributing to innate immunity.
Autophagy and human immunodeficiency virus 1 (HIV1): the role of the viral Tat proteins
To their overall growth advantage, some viruses are known to suppress autophagy, which supports the view that autophagy is an important mechanism of host defense. The role of autophagy in anti-HIV innate immunity is, however, not well defined and requires clarification. For example, HIV Nef binds to Beclin1 (ATG6) and inhibits autophagy; on the other hand, HIV replication is enhanced by autophagy, suggesting that autophagy both promotes and antagonizes HIV infection. A recent paper reports that HIV infection activates the mTOR pathway and inhibits autophagy in dendritic cells, indicating that some viruses inhibit autophagy as a strategy to evade innate immune responses.
HIV-1 encodes the transactivator Tat, which is synthesized early in infection and stimulates viral transcription. Tat proteins are also secreted from infected cells and act as chemokines to influence the motility of neutrophils and macrophages. Moreover, Tat proteins are capable of passing through the cell membrane and penetrating neighboring uninfected cells. Accumulating evidence indicates that, when they penetrate uninfected monocytes and macrophages, Tat proteins profoundly affect the innate immune responses. HIV-1 strains prevalent in Africa and India carry Tat C, which differs in several amino acids from Tat B in the HIV-1 strains common in western countries, including the USA. The goal of this project is to elucidate the distinct immunoregulatory activity of the two Tat proteins in human monocytes and macrophages. To this end, we performed quantitative RT–PCR analysis to test the effect of recombinant Tat B and Tat C on the expression of various cytokine/chemokine genes in human peripheral monocytes. We found that both Tat proteins induce various cytokines and chemokines, such as type I interferons, IL-12p40, IL6, CXCL10, HuMIG, and ISG15, in a differential manner in monocytes. In order to establish an alternative route of Tat protein entry, we constructed flag-tagged Tat B and Tat C and expressed them in THP-1 human monocyte–like cells, as well as in primary monocyte–derived human MФ, and began studying their effects on gene expression patterns.
Histone methyltransferase WHSC1 directs H3.3 incorporation in activated genes by recruiting the chromatin assembly factor HIRA.
H3.3 is very similar to the canonical histone H3 (H3.1 and H3.2) in its 3D structure, differing only in a few amino acids. Unlike the canonical histone H3, however, H3.3 (encoded by two genes) is synthesized outside the S phase and is incorporated into chromatin during transcription. It is thought that H3.3 associates with the preexisting canonical histone H4 to form a new nucleosome, presumably along with H2AZ and the preexisting H2B. H3.3 incorporation into chromatin requires specific chromatin assembly factors distinct from those involved in replication-coupled histone deposition. HIRA is a major H3.3–specific histone chaperone dedicated to transcription-coupled H3.3 incorporation. HIRA carries the WD40 domain and B domain, through which it interacts with three other subunits to act as a chromatin-assembly factor. Another H3.3–specific chaperone, ATRX, is reported to be involved in H3.3 incorporation into the telomeres.
We are interested in transcription coupled H3.3 incorporation. During the past year, we studied the mode and kinetics of H3.3 deposition during and after transcriptional activation. Analyzing the interferon (IFN)–stimulated genes (ISGs) Ifit1, Mx1, Oas1, and Stat1 as a model, we showed that H3.3 is incorporated into all four ISGs as soon as their transcription begins. The notable features of IFN–induced H3.3 deposition were that H3.3 deposition displayed a sharp positional gradient, in that the deposition was the greatest in the 3′ transcription end site, and that H3.3 deposition continued long after transcription had ceased, even 48 h after IFN stimulation. Essentially, an identical pattern of H3.3 deposition was seen for the c-Fos and c-Jun genes, which encode proteins that form the heterodimeric transcription factor AP-1 after UV stimulation. We noted that trimethylation of H3K36 was induced after stimulation and that the temporal and positional patterns of increase closely correlated with H3.3 incorporation. Methylation of H3K36 is catalyzed by the SET2 family of histone methylases, including WHSC1 (also known as NSD2). We tested whether WHSC1 plays a role in transcription-coupled H3.3 deposition by testing Whsc1–/– mouse embryonic fibroblasts (MEFs). We found that IFN-induced H3.3 deposition was completely absent in Whsc1–/– MEFs, while Whsc1+/+ MEFs supported the expected H3.3 deposition. We found that WHSC1 interacts with the chromatin-binding factor BRD4 and is thus critical for the recruitment of the transcription elongation factor P-TEFb. In the absence of WHSC1, ISG elongation was strongly impaired, resulting in severe reduction of ISG mRNA induction. Supporting the role of WHSC1 in elongation, the elongating form of RNA polymerase II (S2Pol II) was present on the ISGs at very low levels in Whsc1–/– cells. Chromatin immunoprecipitation (ChIP) analysis demonstrated that WHSC1 was recruited to the ISGs upon IFN stimulation. Distributed throughout the coding regions, WHSC1 remained on the ISGs long after the cessation of ISG transcription. In parallel with WHSC1 recruitment, HIRA bound to the ISGs after IFN stimulation and remained on ISGs as long as did WHSC1. Furthermore, WHSC1 directly interacted with HIRA through the internal PHD domain and the HMG domain independently of the interaction with BRD4. Consistent with the interaction of WHSC1 and HIRA, WHSC1 and HIRA bound to the ISGs as a protein complex, as confirmed by re-ChIP assays. Taken together, our studies reveal two phases of H3.3 deposition: an initial elongation-coupled phase of deposition, which is then switched to the post-transcription phase of deposition. It is clear that WHSC1 plays a central role in both phases.
Construction of H3.3–HA knock-in mice and validation of the experimental model
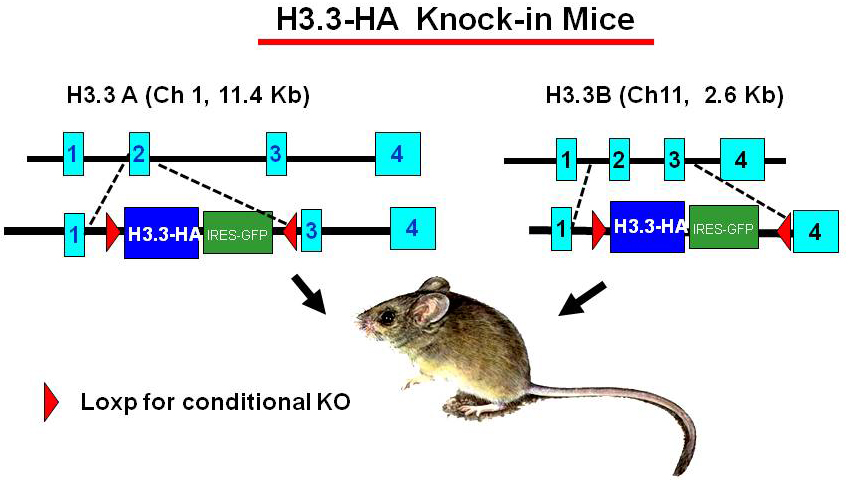
Click image to enlarge.
Figure 1. H3.3–HA knock-in mice
To study the activity of H3.3 in vivo, we generated mouse strains in which the endogenous H3.3 genes (H3.3A, H3f3a and H3.3B, H3f3b) are replaced by hemagglutinin (HA)-tagged H3.3 genes (Figure 1). The knock-in targeting vectors contain HA-tagged H3.3 cDNA (blue) and an IRES–driven GFP (green) to allow independent monitoring of the expression of the inserted genes. The inserted H3.3A–HA and H3.3B–HA fragments are flanked by the LoxP sites (red) to allow conditional knockout of the genes. The original targeting vectors had the neomycin- and hygromycin-resistant genes for ES cell selection, and were removed by FRT–based recombination to avoid potential adverse effects in the mouse. Heterozygous mice for the H3.3A–HA and H3.3B–HA loci are viable and fertile, although the birth rate of homozygous mice appears significantly lower than that expected for Mendelian inheritance. We confirmed that H3.3–HA are expressed at variable levels throughout adult tissues. Preliminary ChIP analysis performed with anti–HA antibody indicated that H3.3–HA is deposited in induced genes in bone marrow–derived macrophages stimulated with IFNg and toll-like receptor (TLR) signaling. Mouse embryonic fibroblasts (MEFs) that carry homozygous and heterozygous H3.3A–HA and H3.3B–HA have been established. We plan to analyze the macrophages and MEFs for global H3.3 deposition using the ChIP-seq approach.
Additional Funding
- Trans NIH-FDA Biodefense Program, IATAP Program
Publications
- Nishiyama A, Dey A, Tamura A, Ko, M, Ozato K. Activation of JNK triggers release of Brd4 from mitotic chromosomes and mediates protection from drug-induced mitotic stress. PLoS One 2012;7(5):e34719.
- Miyagawa F, Zhang H, Terunuma A, Ozato K, Tagaya Y, Katz SI. IRF8 as a critical determinant in CD8 T cell effector function. Proc Natl Acad Sci USA 2012;109:12123-12128.
- Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, Takami M, Ichino M, Nakazawa M, Matsuyama T, Kamijo R, Kitagawa S, Ozato K, Tamura T. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS One 2011;6(10):e25812.
- Devaiah B, Lewis B, Mertz J, Cherman N, Robey P, Ozato K, Sims R, Singer D. BRD4 is a atypical kinase that phosphorylates Ser2 of the RNA Pol II C-terminal domain. Proc Natl Acad Sci USA 2012;109:6927-6932.
- Chang T-H, Xu S, Tailor P, Kanno T, Ozato K. The SUMO deconjugating enzyme SENP1 switches IRF8 from a repressor to an activator and promotes macrophage activation. J Immunol 2012;189:3548-3556.
Collaborators
- Toru Kubota, PhD, Japan National Institute of Infectious Diseases, Tokyo, Japan
- Ben-Zion Levi, PhD, Technion, Israel Institute of Technology, Haifa, Israel
- Herbert Morse II, MD, Laboratory of Immunopathology, NIAID, Rockville, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
Contact
For more information, email ozatok@mail.nih.gov or visit ozatolab.nichd.nih.gov.