

You are here: Home > Section on Human Genetics
The Chromatin-Based Epigenome in Development and Aging

- Bruce H. Howard, MD, Head, Section on Human Genetics
- Valya Russanova, PhD, Staff Scientist
- Irene Stevens, BS, Postbacccalaureate Intramural Research Training Award Student
The normal human lifespan is marked by a complex series of developmental transitions, with relative stability during adulthood and, ultimately, a gradual decline in viability. Clock-like mechanisms presumably underlie the developmental events that occur through childhood and adolescence. Further, instabilities in such mechanisms are likely to be an integral part of the aging process and to contribute to many common degenerative diseases of later life. A breakthrough in the area of developmental transitions would have remarkable implications and underscores today's widespread interest in the rapidly evolving field of epigenomics as the key to further progress.
Whole genome surveys of epigenome structure
The past year saw continued application of next-Gen sequencing approaches to studies that use human tissues from several clinical sources. We obtain peripheral blood monocytes from newborns (cord blood) through a collaboration with the Perinatology Branch, NICHD, while monocytes from adults are available through the NIH Department of Transfusion Medicine. The cells are analyzed either directly or after differentiation in vitro into antigen-presenting dendritic cells. We procure human skin fibroblasts from newborns and adults, as needed, under a protocol approved by the NICHD Institutional Review Board. Third, through collaborative work with Alan DeCherney, we obtain human ovarian granulosa cells harvested in association with ART (assisted reproductive technology) services at Shady Grove Hospital (Gaithersburg, MD).
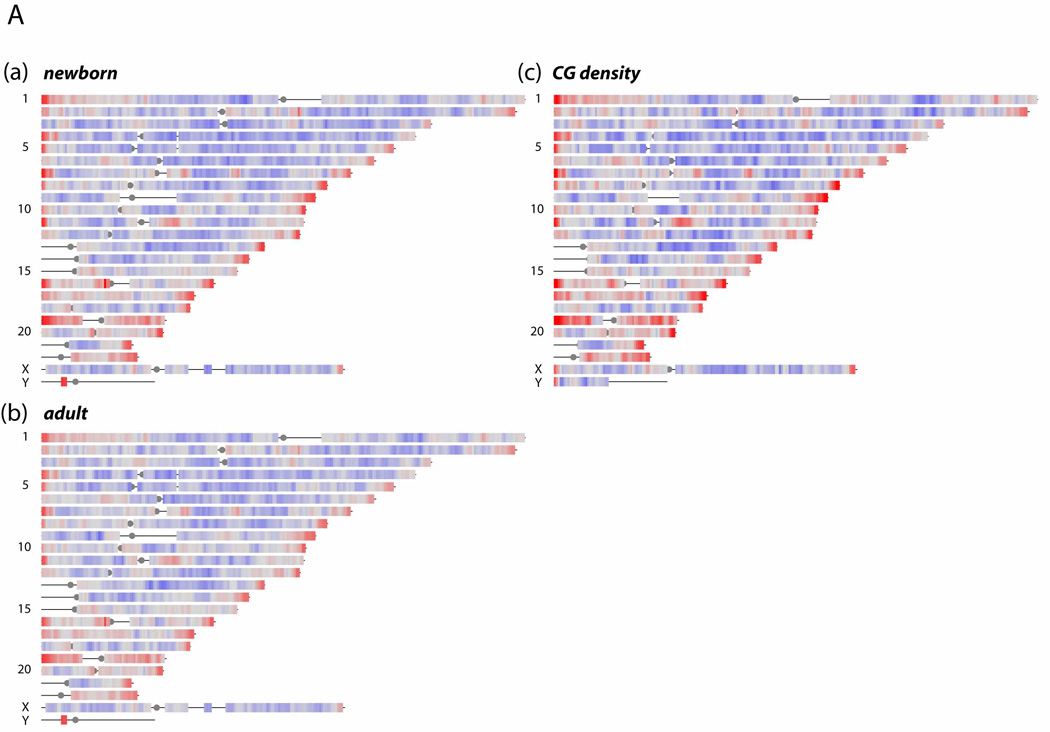
Click image to enlarge.
Figure 1. DNA methylation levels on a whole-genome scale
Sequence reads from precipitated MeDIP DNA were analyzed with a 2 Mb genomic window (1 Mb steps) and aligned to single-copy (RepeatMasker excluded) regions of the human genome, build 36.1. Red color represents ≥ 2-fold over-representation, and blue ≤ 2-fold under-representation for MeDIP signals compared with input control DNA. (c) CG dinucleotide densities in single-copy regions relative to genome average.
With both monocyte- and fibroblast-based experimental systems, we previously employed microarrays to study developmental and age-related changes in gene regulation. Recent experiments with granulosa cells used RNA-seq to characterize gene expression. Our current work focuses on whole-genome surveys to explore the epigenome features that underlie changes in gene function. In mammalian cells, cis-dependent epigenetic states are, of course, maintained by both chromatin structure and DNA methylation. We assess chromatin states with respect to histone acetylation and methylation patterns, as well as the topologies of these patterns extending over domains that typically encompass one or more genes. We characterize DNA methylation at the whole genome level (MeDIP-seq, MethylCap-seq), as well as at nucleotide resolution over gene-size domains (for the latter we have optimized the rapid design and testing of tiled, nested primer pairs suitable for bisulfite-modified DNA). Of particular interest are interactions between the chromatin- and DNA methylation–based components of epigenetic control.
Given the rapid progress of epigenomics and the very large data sets generated by chromatin immunoprecipitation (ChIP) combined with next-Gen sequencing–based (ChIP-seq) analyses, continuing refinement of bioinformatics tools is essential. Along these lines, we constantly improve and extend our genome annotation, pattern recognition, and pattern comparison algorithms. For topology-based mapping of chromatin patterns, we are also collaborating with the Section on Chromatin and Gene Expression (headed by David Clark) on the small yeast genome, which permits much deeper sequencing coverage, and thus much higher resolution patterns. Finally, new bioinformatic tools are being developed to efficiently link patterns in RNA expression data sets with epigenome features.
Our chromatin-related results to date indicate that genes subject to both differentiation and developmental controls depend on the three-dimensional topology of the genome and are sensitive to the remodeling of higher-order chromatin structures. We will continue to focus on large (on the order of 100 kb) domains over which histone acetylation or histone H3–K27 patterns are altered, as well as on control elements that may assume non–B DNA conformations. DNA methylation–related results indicate that a little-studied subcompartment of the genome consisting of small sets of regions with high homology—either within gene clusters or dispersed gene families—may be substantially enriched for post-natal developmental- and age-related epigenome remodeling. If our hypothesis is correct, such regions may provide clock-like functions of considerable importance.
The emerging goal is to generalize our paradigm of dynamic postnatal epigenome structure to address a range of current problems in maternal reproductive health, pediatrics, and areas of medicine relating to age-related disease. Based on the genes currently under study, we will most likely investigate deficiencies in the innate immune systems of newborns, peripheral insulin resistance and diabetes in adolescents and young adults, and a spectrum of neurodegenerative processes, including Parkinson's and Alzheimer's diseases, in the elderly.
Additional Funding
- Supplementary funding from the Program in Reproductive and Adult Endocrinology, NICHD
- Scientific Director's challenge award
Publications
- Cole HA, Howard BH, Clark DJ. The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc Natl Acad Sci USA 2011;108:12687-12692.
- Cole HA, Howard BH, Clark DJ. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res 2011;39:9521-9535.
- Salpea P, Russanova V, Hirai T, Sourlingas TG, Sekeri-Pataryas KE, Romero R, Epstein J, Howard BH. Postnatal development- and age-related changes in DNA methylation patterns in the human genome. Nucleic Acids Res 2012;40:6477-6494.
Collaborators
- David J. Clark, PhD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
- Alan DeCherney, MD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- Jonathan Epstein, MS, Molecular Genomics Laboratory, NICHD, Bethesda, MD
- Roberto Romero, MD, Program in Perinatal Research and Obstetrics, NICHD, Detroit, MI
- James Segars, MD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
Contact
For more information, email howard@helix.nih.gov.