

You are here: Home > Section on Genomic Imprinting
Molecular Genetics of an Imprinted Gene Cluster on Mouse Distal Chromosome 7

- Karl Pfeifer, PhD, Head, Section on Genomic Imprinting
- Bokkee Eun, PhD, Visiting Fellow
- Claudia Gebert, PhD, Research Fellow
- Megan Sampley, PhD, Postdoctoral Fellow
- Austin Good, BS, Postbaccalaureate Fellow
- Cameron Johnson, BS, Postbaccalaureate Fellow
Genomic imprinting is an unusual form of gene regulation by which an allele's parental origin restricts allele expression. Imprinting is critical for normal development, and recent studies have especially emphasized a role for imprinting in normal brain development and function (1). Imprinted genes are not randomly scattered throughout the chromosome but rather are localized in discrete clusters. One cluster of imprinted genes is located on the distal end of mouse chromosome 7 (Figure 1). The syntenic region in humans (11p15.5) is highly conserved in gene organization and expression patterns. Mutations disrupting the normal patterns of imprinting at the human locus are associated with developmental disorders and many types of tumors. In addition, inherited cardiac arrhythmia is associated with mutations in the maternal-specific Kcnq1 gene. We use mouse models to address the molecular basis for allele-specific expression in this distal 7 cluster. We use imprinting as a tool to understand the fundamental features of epigenetic regulation of gene expression. We also generate mouse models for the several inherited disorders of humans. We have generated models to study defects in cardiac repolarization associated with loss of Kcnq1 function and, more recently, characterized the phenotype associated with loss of Calsequestrin2 gene function.
Molecular basis for allele-specific expression of the mouse H19 and Igf2 genes
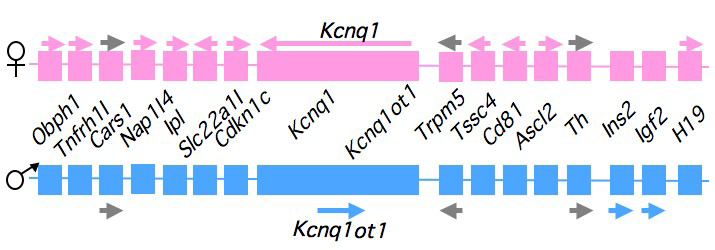
Figure 1. An imprinted domain on mouse distal chromosome 7
Maternal (pink) and paternal (blue) chromosomes are indicated. Horizontal arrows denote RNA transcription.
Our studies on the mechanisms of genomic imprinting focus on the H19 and Igf2 genes, which lie at one end of the distal 7 imprinted cluster (Figure 1). Paternally expressed Igf2 lies about 80 kb upstream of the maternal-specific H19 gene. Using cell culture systems as well as transgene and knockout experiments in vivo, we identified the enhancer elements responsible for activation of these two genes. The elements are largely shared and located downstream of the H19 gene. Parent-of-origin–specific expression of both genes is dependent on a 2.4 kb shared element (called the H19ICR for H19 imprinting control region [ICR]) located just upstream of the H19 promoter and thus juxtaposed between the Igf2 gene and the shared enhancers. The CpG sequences within this element are methylated specifically on the paternally inherited chromosome. Our conditional ablation of this element in vivo demonstrates that the non-methylated H19ICR—i.e., the copy on the maternal chromosome—is continually required for silencing of the maternal Igf2 allele. Knockin experiments demonstrated that the H19ICR contains a methylation-sensitive transcriptional insulator. Thus, on the non-methylated maternal chromosome, the active insulator within the H19ICR prevents activation of Igf2 by the downstream enhancers. Methylation of the paternal chromosome inactivates the insulator and permits Igf2 expression. Unexplained by this model is the effect of several small differentially methylated regions (DMRs) proximal to the Igf2 transcription unit. We are currently investigating the mechanistic significance of these elements. Imprinting of H19 occurs via a distinct genetic mechanism. The conditional ablation of the H19ICR indicates that it is not continuously required to silence the paternal allele. Rather, the H19ICR is required early in development to establish a novel epigenetic state at the H19 promoter. Once in this novel epigenetic configuration, the H19 promoter is stably refractory to transcriptional activation. Current studies indicate that the epigenetic program includes, but is not solely limited to, the hypermethylation of the H19 promoter.
To determine which elements are necessary and sufficient for imprinting at the locus, we moved the H19ICR and its mutated derivatives to heterologous loci. Our results demonstrated that the ICR alone is sufficient to imprint a normally non-imprinted chromosome. Moreover, such activity does not depend on germline differences in ICR methylation (2). Thus, the ICR likely marks its parental origin by a mechanism that is independent of DNA methylation. To determine the epigenetic signals that constitute the genomic imprint, we are now conducting genetic and molecular analyses of primordial germ cells isolated from wild-type and mutant mice.
One important result from the last year is our identification of a long non-coding RNA, coincident with the mesodermal enhancer, that acts as a rheostat to regulate activation levels of that enhancer.

Figure 2. Distinct maternal and paternal chromosomal conformations at the distal 7 locus
Epigenetic modifications on the 2.4 kb ICR generate alternative 3D organizations across a large domain on paternal (blue) and maternal (pink) chromosomes and thereby regulate gene expression. ICR, imprinting control region; EE, endodermal enhancer; ME, mesodermal enhancer.
We are also continuing a series of experiments to elucidate the molecular mechanisms by which the H19ICR may act as a transcriptional insulator. Several groups demonstrated four CTCF–binding sites within the H19ICR. CTCF (CCCTC–binding factor—a zinc finger protein) is a DNA–binding protein previously demonstrated to interact with the chicken beta-globin insulator. The ability of CTCF to recognize DNA is sensitive to methylation, i.e., CTCF cannot bind to the methylated paternally inherited ICR, thus explaining the activation of the paternal Igf2 allele. To understand the molecular basis for insulator function, we conducted a series of experiments to characterize the three-dimensional organization of the Igf2/H19 locus, comparing maternal and paternal and wild-type and mutant chromosomes (Figure 2). We demonstrated that transcriptional activation is invariably associated with physical interaction of the promoter and enhancer elements. Insertion of the ICR/insulator abrogates these interactions and induces alternative long-range chromosomal interactions between it and the regulated promoter elements. Curiously, induction of these alternative interactions with the insulator is enhancer-dependent.
Our research goals for the next year call for (i) identifying the original gametic imprint that distinguishes the parental origin of chromosome 7; (ii) determining the molecular mechanisms that convert the gametic imprint into parent-of-origin–specific patterns of DNA methylation; (iii) identifying key transcription factors associated with the Igf2 and H19 promoters; (iv) comparing promoter structure on wild-type and mutant chromosomes, both maternal and paternal, in order to determine the molecular role of enhancer and insulator elements; and (v) characterizing the molecular mechanisms of the shared H19 and Igf2 enhancers. Specifically, we will use genetic and molecular approaches to characterize wild-type and mutant chromosomes to identify the role of enhancer-associated non-coding RNAs.
Mouse models for inherited long QT syndrome
Inherited long QT syndrome (LQTS) is characterized by an abnormal electrocardiogram indicative of repolarization defects; it can result in syncope or sudden death. Romano-Ward syndrome (RWS) patients generally inherit the LQTS disorder as a dominant phenotype and show no other traits. Jervell and Lange-Nielsen syndrome (JLNS) patients display profound congenital deafness in addition to LQTS. Both phenotypes are recessive. We generated several mutations in the mouse Kcnq1 gene to model the human diseases (3). Ablation of the gene results in vestibular and auditory defects. Histological analyses suggested that the defects are attributable to deficiency in the K+ recycling pathway that is crucial for generating endolymph, the specialized fluid bathing the inner hair cells. ECG tracings of mutant mice indicate profound defects in cardiac repolarization when measured in vivo. However, the defects are not evident in isolated hearts ex vivo, indicating that the KCNQ1 protein plays a key role in mediating critical extracardiac signals. Further analyses demonstrated that KCNQ1 function is specifically required to modulate cardiac function in the presence of beta-adrenergic stimulation.
Biochemical and pharmacological studies both predicted that the key biological function of the KCNQ1 protein is to associate with the helper protein KCNE1 to form the IKS potassium channel. One of the most novel results of our studies is the discovery that ablation of the Kcnq1 gene leads to cardiac defects in addition to those noted in Kcne1–deficient mice. The results demonstrated a novel role for KCNQ1 in heart development and/or function. We used our mutant mice as tools to detect a previously unappreciated potassium channel that was dependent on KCNQ1 but not on KCNE1. We are now investigating the role of this channel in mouse and human hearts.
Recent human genetics studies indicate that KCNQ1 polymorphisms are associated with increased risk for diabetes. Our analysis demonstrates that mice lacking functional Kcnq1 show enhanced insulin sensitivity (3).
The role of calsequestrin2 in regulating cardiac function
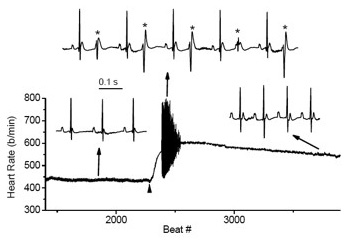
Figure 3. Cardiac arrhythmias in calsequestrin-2 deficient mice phenocopy the human disease.
Premature ventricular complexes (*) are induced by stress in Casq2-deficient but not in wild-type mice.
Mutations in the CASQ2 gene, which encodes cardiac calsequestrin (CASQ2), are associated with catecholaminergic polymorphic ventricular tachycardia (CPVT) and sudden death. The survival of individuals homozygous for loss-of-function mutations in CASQ2 is surprising, given the central role of Ca2+ ions in excitation-contraction (EC) coupling and the presumed critical roles of CASQ2 in regulating Ca2+ release from the sarcoplasmic reticulum (SR) into the cytoplasm. To address this paradox, we generated a mouse model for loss of Casq2 gene activity. Comprehensive analysis of cardiac function and structure generated several important insights into CASQ2 function (4). First, CASQ2 is not essential to provide sufficient Ca2+ storage in the SR of the cardiomyocyte. Rather, a compensatory increase in SR volume and surface area in mutant mice appears to maintain normal Ca2+ storage capacity. Second, CASQ2 is not required for the rapid, triggered release of Ca2+ from the SR during cardiomyocyte contraction. Rather, the RyR receptor opens appropriately, resulting in the normal, rapid flow of Ca2+ into the cytoplasm, thus allowing normal contraction of the cardiomyocyte. Third, CASQ2 is required for normal function of the RyR during cardiomyocyte relaxation. In the absence of CASQ2, significant Ca2+ leaks occur through the RyR and lead to premature contractions and cardiac arrhythmias (Figure 3). Fourth, CASQ2 function is required to maintain normal junctin and triadin levels. We do not yet understand what role, if any, the compensatory changes in these two SR proteins play in modulating the loss-of-Casq2 phenotype.
To address these issues and to model cardiac disorders associated with late-onset (not congenital) loss of CASQ2 activity, we established and are analyzing two new mouse models in which changes in the CASQ2 gene structure are induced by hormone treatment. In the first model, an invested/null allele is restored to normal function by the addition of hormone. In the past year, we demonstrated the effectiveness of this model and noted that full Casq2 protein levels are restored within one week of treatment. In the second model, a functional gene is ablated by the addition of hormone. The Casq2 gene and mRNAs are deleted from cardiac cells within 4 days of hormone treatment. Phenotypic analyses shows that these mice present a profoundly more defective heart than congenitally mutant mice; that is, arrhythmias are more frequent, of longer duration, and qualitatively are much more severe. Our results indicate that the developmental program has flexibility in coping with the loss of Casq2 protein that the adult heart lacks. Our current efforts focus on finding the molecular basis for the coping mechanisms as a means to better understand cardiac development and to perhaps identify clues as to how to handle Casq2 deficiency that may be of therapeutic value.
Additional Funding
- NIH Office of Education
Publications
- Cunningham MD, Kassis J, Pfeifer K. Chromatin modifiers, cognitive disorders, and imprinted genes. Dev Cell 2010;18:169-170.
- Gebert CM, Kunkel D, Grinberg A, Pfeifer K. H19 imprinting control region methylation requires an imprinted environment only in the male germ line. Mol Cell Biol 2010;30:1108-1115.
- Boini KM, Graf D, Hennige AM, Koka S, Dempe DS, Wang K, Ackerman TF, Foeller M, Vallon V, Pfeifer K, Schleiber E, Ullrich S, Haering HU, Haeussinger D, Lang F. Enhanced insulin sensitivity of gene-targeted mice lacking functional KCNQ1. Amer J Physiol Regul Integr Comp Physiol 2009;296:R1695-1701.
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 2007;116:2510-2520.
- Ebert SN, Rong Q, Boe S, Pfeifer K. Catecholamine synthesizing cells in the embryonic mouse heart. Annals NY Acad Sci 2009;1148:317-324.
Collaborators
- Steve Ebert, PhD, Burnett College of Biomedical Sciences, University of Central Florida, Orlando, FL
- Bjorn Knollmann, MD, PhD, Vanderbilt University Medical Center, Nashville, TN
- Florian Lang, MD, Universität Tübingen, Tübingen, Germany
Contact
For more information, email kpfeifer@helix.nih.gov or visit pfeiferlab.nichd.nih.gov.