Biophysics of Large Membrane Channels

- Sergey M. Bezrukov, PhD, DSci, Head, Section on Molecular Transport
- Tatiana K. Rostovtseva, PhD, Associate Scientist
- María Queralt-Martín, PhD, Visiting Fellow
- Megha Rajendran, PhD, Visiting Fellow
- William M. Rosencrans, BS, Intramural Research Training Award Fellow
The Section studies mechanisms of channel-facilitated transport across cell and organelle membranes by combining experiments on channel reconstitution into planar lipid membranes with the physical theory, allowing for both qualitative and quantitative understanding of the transport processes. We focus mostly on large, metabolite-transporting channels such as VDAC (voltage-dependent anion channel from the outer membrane of mitochondria), OmpF (general bacterial porin from Escherichia coli), LamB (sugar-specific bacterial porin from Escherichia coli), OprF (porin from Pseudomonas aeruginosa), MspA (major outer-membrane porin from Mycobacterium smegmatis), alpha-Hemolysin (toxin from Staphylococcus aureus), and translocation pores of Bacillus anthracis (PA63), Clostridium botulinum (C2IIa), and Clostridium perfringens (Ib) binary toxins, Epsilon toxin (from Clostridium perfringens), Alamethicin (amphiphilic peptide toxin from Trichoderma viride), Syringomycin E (lipopeptide toxin from Pseudomonas syringae), and the bacterial peptide TisB involved in persister cell formation. We also use Gramicidin A (linear pentadecapeptide from Bacillus brevis) as a molecular sensor of membrane mechanical properties.
To investigate these mostly beta-barrel channels under precisely controlled conditions, we first isolate the channel-forming proteins from their host organisms, or use recombinant proteins, and then reconstitute them into planar lipid membranes. Healthy cell functioning and development require effective communication between cells and cell organelles, which is facilitated by membrane channels. Our main goal is to elucidate the physical principles of regulation of such channels under normal and pathological conditions. Among many wet-lab approaches, such as fluorescence correlation spectroscopy, bilayer overtone analysis, and confocal microscopy, single-channel electrophysiology is our hallmark technique that allows us to study transport processes at the single-molecule level. In our latest projects, we combine biophysical methods with those of cell biology. Empirical findings obtained in experiments are rationalized within the framework of a physical theory of channel-facilitated transport, which brings an understanding necessary to design new strategies to effectively correct the deviant interactions associated with disease.
Molecular mechanism of neuroprotective drug action through targeting alpha-synuclein interaction with mitochondrial VDAC
The second most common neurodegenerative disorder in the U.S. is Parkinson’s disease (PD), a neurological condition that causes severe movement problems. It is well known that the disease is associated with abnormal accumulation of a protein called alpha-synuclein, which is naturally produced in healthy neurons. Previous studies have shown that, when alpha-synuclein builds up in cells, it somehow moves through the mitochondrial outer membrane and targets respiratory complexes at the inner membrane, inducing mitochondrial dysfunction. However, the gateway that alpha-synuclein uses to enter mitochondria has long been a mystery. A hint to this puzzle appeared several years ago when our lab, using in vitro experiments with reconstituted mitochondrial VDAC, found that alpha-synuclein can translocate through this channel. This year, working in collaboration with Parkinson’s disease researchers in the lab of Mark Cookson, we showed that VDAC is indeed a pathway for alpha-synuclein translocation into the mitochondria in cells. In this work, we studied effects of a cholesterol-like synthetic compound called olesoxime, which was previously shown to defend mitochondria from the alpha-synuclein–induced toxicity. Olesoxime generated considerable interest for its ability to protect neurons and mice in a range of neurodegenerative conditions, including PD. It was also established that olesoxime binds to VDAC in cells.
Following our interest in alpha-synuclein interaction with VDAC, we undertook to understand whether the molecular mechanism of olesoxime neuroprotection involves the interaction. Using neuronally differentiated human cells overexpressing wild-type alpha-synuclein as a cell model of PD, we found that olesoxime inhibits alpha-synuclein translocation into mitochondria. By applying a set of complementary electrophysiological and biophysical approaches, we provided mechanistic insights into the interplay between alpha-synuclein, VDAC, and olesoxime. Consistent with past studies, we found that overexpressed alpha-synuclein induces cell death and that olesoxime treatment dramatically reduced its rate by keeping mitochondria healthy. Working at the single-molecule level, from the data obtained in channel-reconstitution experiments, we deduced that olesoxime interacts with the VDAC beta-barrel at the lipid-protein interface in such a way that it hinders alpha-synuclein translocation through the VDAC pore and affects VDAC voltage gating. The findings suggest that the alpha-synuclein interaction with VDAC is a new target for the development of drugs against alpha-synuclein toxicity. In particular, the use of molecules interacting with the alpha-synuclein–VDAC complex could be a promising and effective pharmacological treatment for a wide range of neurodegenerative conditions, such as spinal muscular atrophy, amyotrophic lateral sclerosis, Parkinson’s and Huntington’s diseases, and chemotherapy side effects, aimed to decrease mitochondrial deficiencies in affected neurons.
Figure 1. Proposed model of the olesoxime neuroprotective effect
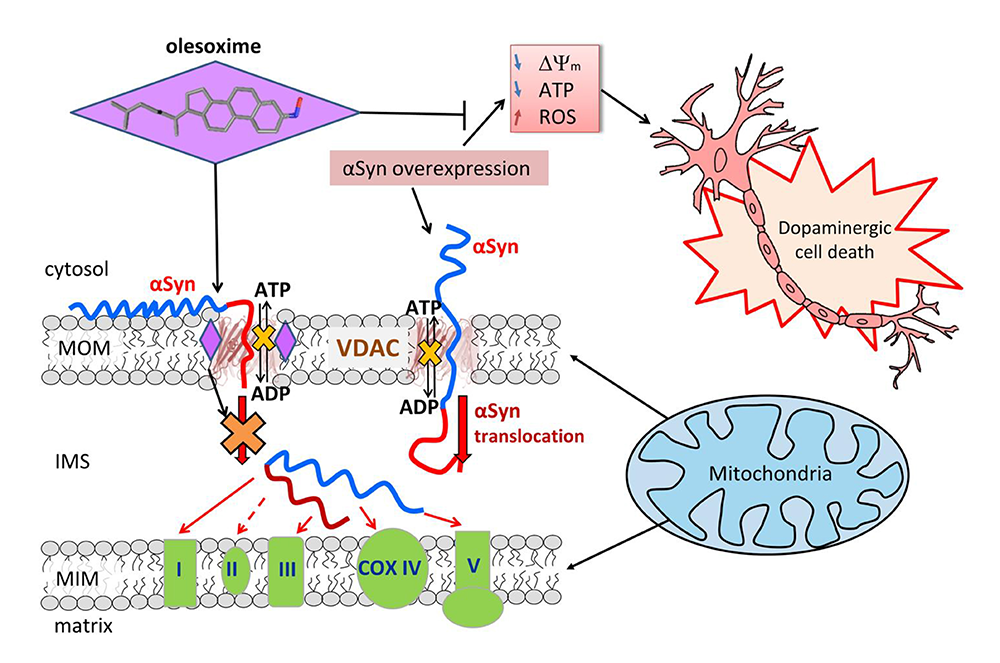
Click image to view.
When alpha-synuclein (aSyn) is captured by the VDAC pore, it disrupts ATP/ADP fluxes through the VDAC. Under normal conditions, endogenous aSyn regulates the fluxes by reversibly and dynamically blocking the VDAC. Stress resulting from aSyn overexpression during Parkinson’s disease induces aSyn translocation across the mitochondrial outer membrane (MOM) via the VDAC and targets electron transport chain complexes (ETC) in the inner membrane (MIM), causing their impairment, mitochondrial dysfunction, and eventually neuronal cell death. Olesoxime partitions into the MOM and hinders aSyn translocation through the VDAC by interacting with the pore-lipid interface. The model suggests a tentative mechanism of olesoxime protection of mitochondria integrity and promotion of neuronal cell survival.
Effect of a post-translational modification mimic on protein translocation through a nanopore
Post-translational modifications (PTMs) of proteins are recognized as crucial components of cell-signaling pathways by modulating folding, altering stability, changing interactions with ligands, and therefore serving many regulatory functions. PTMs occur as covalent changes in the protein’s amino acid side chains or the length and composition of their termini. Inspired by the importance of PTMs in cell signaling, this year we studied the functional consequences of PTMs for alpha-synuclein interactions with mitochondrial VDAC. We mimicked PTMs of the 140–amino-acid cytosolic protein alpha-synuclein by attaching divalent Alexa Fluor 488 to the beginning and end of its C-terminal tail. Each of these modifications increases the total negative charge of the tail by two elementary charges and introduces extra bulkiness at the modification location, significantly changing the dynamics of the alpha-synuclein interaction with the VDAC pore. Using single-channel reconstitution into planar lipid membranes, we found that such PTM–like modifications change interactions drastically in both the efficiency of VDAC inhibition by alpha-synuclein and its translocation through the VDAC pore. We analyzed time-resolved single-molecule events of alpha-synuclein capture by the VDAC pore within a framework of a one-dimensional diffusion model, using an interaction “quasi-potential” that incorporates mostly electrostatic and entropic components of alpha-synuclein interaction with the pore. The analysis proved to be an effective means of quantitatively describing the PTM–like modification effects on the kinetics of the capture, release, and translocation and yielded the positions of the modifications with an excellent precision of about three residues. Notably, the technique is general and can readily be applied to other biological channels and nanopores, suggesting that it could be extended to quantify populations of proteins that have undergone PTMs. Thus, in view of the recently established role of disordered charged termini of cytosolic proteins in the control of VDAC–facilitated transport, our findings establish a new mechanism of PTM–induced regulation of protein function.
Figure 2. Probing alpha-synuclein modifications with a VDAC nanopore
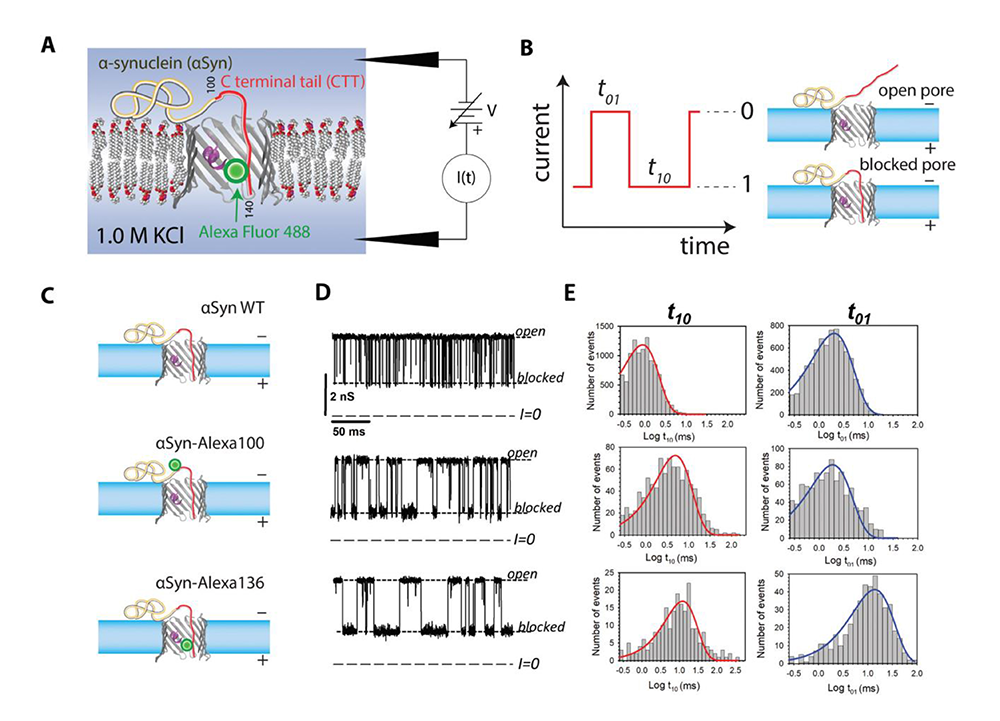
Click image to view.
A. Experimental setup (not to scale): the acidic C-terminal tail (CTT) of membrane-bound aSyn is drawn into the nanopore by an externally applied transmembrane voltage.
B. Definitions of open (0) and blocked (1) states; durations in each state are denoted t01 and t10, respectively.
C. Schematic of three aSyn constructs: the WT, and aSyn labeled with Alexa Fluor 488 at residues 100 and 136, aSyn-Alexa100 and aSyn-Alexa136, respectively.
D. Experimentally observed stochastic fluctuations of single VDAC pore conductance between open and blocked states for three aSyn constructs at −30 mV applied voltage; dashed lines indicate VDAC open and aSyn-blocked states and zero current.
E. Corresponding to the traces in D, distributions of state t01 and t10 durations show quantitative differences in the kinetics of the aSyn–nanopore interaction introduced by Alexa Fluor 488 functionalization.
Blocker escape kinetics from a membrane channel analyzed by mapping blocker diffusive dynamics onto a two-site model
Understanding the blockage of ion channels in biological membranes by natural and synthetic compounds is important from both theoretical and practical points of view. On the one hand, the phenomenon of blockage gives a rich example of the behavior of a solute molecule in strong confinement involving multiple interactions with the protein residues lining the channel walls and, in the case of a charged molecule, with the transmembrane electric field. On the other hand, its practical value is clear from the fact that about 13% of all present drugs of the world pharmacopeia act as ion-channel modifiers, with many of them working as channel blockers. This year, we developed an analytical theory of the blocker escape kinetics from the channel, assuming that a charged blocking molecule cannot pass through a constriction region (bottleneck). If the molecule spends a sufficiently long time in the channel, individual blockades in ionic current can be resolved in single-channel experiments. We focused on the effect of the external voltage bias on the blocker survival probability and characteristic times in the channel. The bias creates a potential well for the charged blocker, with the minimum located near the channel bottleneck. When the bias is strong, escape from the channel is a slow process, which allows for time-resolved observation of individual blocking events. We performed our analysis in the framework of a two-site model of the blocker dynamics in the channel. Importantly, the rate constants, fully determining this model, were derived from a more realistic continuum diffusion model. This was done by mapping the latter onto its two-site counterpart which, while being much simpler, captures the main features of the blocker escape kinetics.
Figure 3. Kinetics of blocker escape from a membrane channel
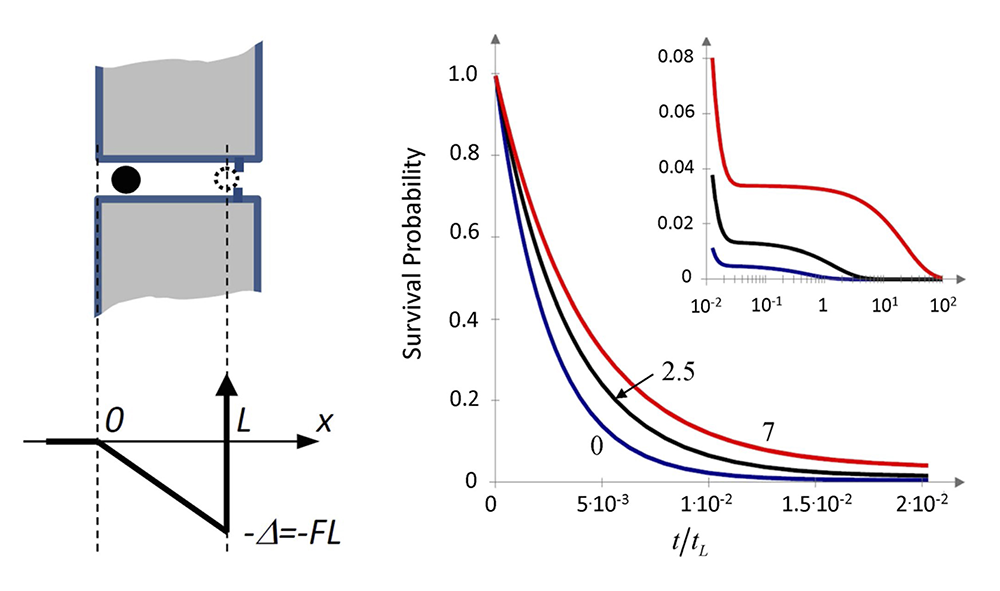
Click image to view.
Left. Cartoon of a blocker in a cylindrical channel and the potential well of depth FL created for the blocker by the biasing force F > 0 in the channel of length L available for the center of the blocking molecule.
Right. Survival probability of the blocker molecule in the channel S(t) at different values of the biasing force and hence the dimensionless well depth increasing from 0 to 2.5 to 7 thermal energy units (lower, middle, and upper curves, respectively) as functions of the dimensionless time; increasing bias results in a slowdown of the decay kinetics of S(t); the inset shows the long-time “tails” of the curves in a finer scale for S(t) and the logarithmic scale for the dimensionless time.
Additional Funding
- Colgate University award to William Rosencrans
Publications
- Rovini A, Gurnev PA, Beilina A, Queralt-Martin M, Rosencrans W, Cookson MR, Bezrukov SM, Rostovtseva TK. Molecular mechanism of olesoxime-mediated neuroprotection through targeting a-synuclein interaction with mitochondrial VDAC. Cell Mol Life Sci 2020;77:3611-3626.
- Rostovtseva TK, Queralt-Martín M, Rosencrans WM, Bezrukov SM. Targeting the multiple physiologic roles of VDAC with steroids and hydrophobic drugs. Frontiers Physiol 2020;11:446.
- Hoogerheide DP, Gurnev PA, Rostovtseva TK, Bezrukov SM. Effect of a post-translational modification mimic on protein translocation through a nanopore. Nanoscale 2020;12:11070-11078.
- Berezhkovskii AM, Bezrukov SM. Blocker escape kinetics from a membrane channel analyzed by mapping blocker diffusive dynamics onto a two-site model. J Chem Phys 2019;150:194103.
- Berezhkovskii AM, Dagdug L, Bezrukov SM. Peculiarities of the mean transition path time dependence on the barrier height in entropy potentials. J Phys Chem B 2020;124:2305-2310.
Collaborators
- Vicente M. Aguilella, PhD, Universidad Jaume I, Castellón, Spain
- Alexandra Beilina, PhD, Cell Biology & Gene Expression Section, NIA, Bethesda, MD
- Alexander M. Berezhkovskii, PhD, Division of Computational Bioscience, CIT, NIH, Bethesda, MD
- Mark R. Cookson, PhD, Laboratory of Neurogenetics, NIA, Bethesda, MD
- Leonardo Dagdug, PhD, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, Mexico
- David Hoogerheide, PhD, National Institute of Standards and Technology, Gaithersburg, MD
- Jennifer C. Lee, PhD, Biochemistry and Biophysics Center, NHLBI, Bethesda, MD
- Ekaterina M. Nestorovich, PhD, The Catholic University of America, Washington, DC
- Sergei Y. Noskov, PhD, University of Calgary, Calgary, Canada
- Olga Protchenko, PhD, Liver Diseases Branch, NIDDK, Bethesda, MD
- Alexander Sodt, PhD, Unit on Membrane Chemical Physics, NICHD, Bethesda, MD
- Gerhard Wagner, PhD, Harvard Medical School, Cambridge, MA
- Michael Weinrich, MD, Office of the Director, NICHD, Bethesda, MD
- David L. Worcester, PhD, National Institute of Standards and Technology, Gaithersburg, MD
- Joshua Zimmerberg, MD, PhD, Section on Integrative Biophysics, NICHD, Bethesda, MD
Contact
For more information, email bezrukov@helix.nih.gov or visit http://smt.nichd.nih.gov.