Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Marianne Knue, CRNP, Nurse Practitioner
- Sara Talvacchio, BSN, Research Nurse
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
- Thanh-Truc Huynh, MS, Biologist
- Suman Goshal, PhD, Postdoctoral Visiting Fellow
- Katerina Hadrava Vanova, PhD, Postdoctoral Fellow
- Abishek Jha, MBBS, Postdoctoral Visiting Fellow
- Ondrej Uher, MSc, Predoctoral Visiting Fellow
- Divya Mamilla, MD, Volunteer
- Matthew Arman Nazari, MD, Volunteer
- Mayank Patel, MD, Volunteer
- Boqun Zhu, MD, Volunteer
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare but clinically important chromaffin-cell tumors that typically arise, respectively, from the adrenal gland and from extra-adrenal paraganglia. The clinical features and consequences of PHEO/PGL, collectively known as PPGLs, result from the release of catecholamines (norepinephrine and epinephrine). An undetected PHEO/PGL poses a hazard to patients undergoing surgery, childbirth, or general anesthesia because of the potential for excess catecholamine secretion, which can result in significant, often catastrophic outcomes. Diagnosing and localizing a PHEO/PGL can be challenging. Plasma and urinary catecholamines, as well as their metabolites, and radio-iodinated metaiodobenzylguanidine (MIBG) scanning can yield false-positive or false-negative results in patients harboring the tumor, and computed tomography (CT) and magnetic resonance imaging (MRI) lack sufficient specificity. The molecular mechanisms by which genotypic changes predispose to the development of PHEO/PGL remain unknown, even in patients with identified mutations. Moreover, in patients with hereditary predispositions, PPGLs differ in terms of their growth, malignant potential, catecholamine phenotype, responses to standard screening tests, various imaging modalities, and therefore to different therapeutic options. We focus on developmental, molecular, genetic, epigenetic, proteomic, metabolomic, immunologic, and other types of studies to investigate the bases for a predisposition to develop PPGLs and the expression of various neurochemical phenotypes and malignant potentials, including therapeutic responses.
Clinical and genetic aspects of pheochromocytoma and paraganglioma
PPGLs are usually benign neuroendocrine tumors. However, PPGLs with mutations in the succinate dehydrogenase B subunit (SDHB) have a poor prognosis and frequently develop metastatic lesions. PPGLs are rare in children, with only a few SDHB mutation–related cases. Previous studies in children were conducted in small cohorts. This large set of pediatric patients provides robust data in the evaluation of clinical outcomes. Thirty-eight males and 26 females were diagnosed with PPGL at a median age of 13 years. The majority of patients displayed norepinephrine hypersecretion, and 73% initially presented with a solitary tumor. Metastases developed in 70% of patients at the median age of 16 years and were mostly diagnosed first two-years and then in years 12–18 post-diagnosis. The presence of metastases at the time of diagnosis had a strong negative impact on survival in males but not in females. The estimated 5-, 10-, and 20-year survival rates were 100%, 97%, and 78%, respectively. We highlighted several important aspects in the management of pediatric patients with SDHB mutation–associated PHEO/PGL. Initial diagnostic evaluation of SDHB–mutation carriers should be started at the age of 5–6 years, with initial work-up focusing on the abdominal region. Thorough follow-up is crucial in the first two years post-diagnosis, and more frequent follow-ups are needed in years 10–20 post-diagnosis because of the increased risk of metastases. Although this age group developed metastasis as early as five years from diagnosis, we showed that the overall 20-year prognosis and survival are good.
A PPGL–related clinical sequela results from elevated catecholamine secretion, which can cause hypertension, tachy-arrhythmia, multi-organ failure, and death. We introduced Ivabradine, a commercially available drug that acts directly on the sinus node in the heart for treatment of severe catecholamine-induced tachy-arrhythmia. We also published some comprehensive reviews on cardiac PGLs, as well as on treatments of arrhythmias.
In another study, we evaluated PPGL patients with the SDHA gene mutation. Our findings suggest that such tumors can occur early and at extra-adrenal locations, behave aggressively, and have a tendency to develop metastatic disease within a short period of time. None of the patients had a family history of PPGL, making them appear sporadic. Nine out of 10 patients showed abnormal PPGL–specific biochemical markers with predominantly noradrenergic and/or dopaminergic phenotype, suggesting their utility in diagnosing and monitoring the disease. A radioconjugate, 68Ga-DOTATATE PET, consisting of the somatostatin analog tyrosine-3-octreotate (Tyr3-octreotate or TATE) labeled with the positron emission tomography (PET) tracer gallium Ga 68 via the macrocyclic chelating agent dodecanetetraacetic acid (DOTA), which may be used as a somatostatin receptor imaging agent in conjunction with PET, was superior to other imaging modalities in localizing these tumors. All seven patients who received conventional therapies (chemotherapy, somatostatin-analog therapy, radiation therapy, 131I-MIBG, peptide-receptor radionuclide therapy) in addition to surgery showed progression.
Brown adipose tissue (BAT) activation is mediated through the action of norepinephrine on β-adrenoceptors (β-ARs). In some malignancies, BAT activation is associated with higher cancer activity. A retrospective case-control study included 342 patients with PPGLs who underwent 18F-fluoro-2-deoxy-D-glucose PET–computed tomography (18F-FDG PET/CT) imaging at the National Institutes of Health (NIH). The presence of active BAT on 18F-FDG PET/CT was associated with lower overall survival than in the control group. The association remained significant after adjusting for the SDHB mutation. Median plasma norepinephrine in the BAT group was higher than the control group. There was a significant association between higher plasma norepinephrine levels and mortality in PPGLs in both groups.
Imaging of pheochromocytomas and paragangliomas
Diverse radionuclide imaging techniques are available for the diagnosis, staging, and follow-up of PPGL. Beyond their ability to detect and localize the disease, the imaging approaches variably characterize the tumors at the cellular and molecular levels and can guide therapy. We updated guidelines jointly approved by the EANM (European Association of Nuclear Medicine) and SNMMI (Society of Nuclear Medicine and Molecular Imaging) for assisting nuclear-medicine practitioners in not only the selection and performance of currently available single-photon emission computed tomography and PET procedures, but also the interpretation and reporting of the results from PPGL patients. We also recently published a review about molecular imaging and radionuclide therapy of PPGL in the era of genomic characterization.
Immune and metabolic aspects of pheochromocytoma and paraganglioma
Therapeutic options for metastatic PHEO/PGL are limited. We therefore tested an immuno-therapeutic approach based on intra-tumoral injections of the antibiotic complex mannan–BAM (biocompatible anchor for membranes) with toll-like receptor ligands (TLRs) into subcutaneous PHEOs in a mouse model (Figure 1). The therapy elicited a strong innate immunity-mediated antitumor response and resulted in a significantly lower PHEO volume compared with the phosphate buffered saline (PBS)–treated group and in a significant improvement in mouse survival. We verified the cytotoxic effect of neutrophils, as innate immune cells predominantly infiltrating treated tumors, in vitro. Moreover, the combination of mannan–BAM and TLRs with agonistic anti–CD40 (CD40 receptors are expressed mainly on antigen-presenting cells such as macrophages and dendritic cells) was associated with increased mouse survival. Subsequent tumor re-challenge also supported adaptive immunity activation, reflected primarily by long-term tumor-specific memory. We verified these results further in metastatic PHEO, where the intra-tumoral injections of mannan–BAM, TLRs, and anti–CD40 into subcutaneous tumors resulted in significantly less intense bioluminescence signals of liver metastatic lesions induced by tail vein injection than in the PBS–treated group. Subsequent experiments focusing on the depletion of T cell subpopulations confirmed the crucial role of CD8+ T cells in the inhibition of bioluminescence signal intensity of liver metastatic lesions. The results call for a new therapeutic approach in patients with metastatic PHEO/PGL by using immunotherapy that initially activates innate immunity, followed by an adaptive immune response.
Figure 1. A novel immunotherapy approach for pheochromocytoma. Reprinted from Uher O, et al. Semin Oncol 2019; 46:385-392.
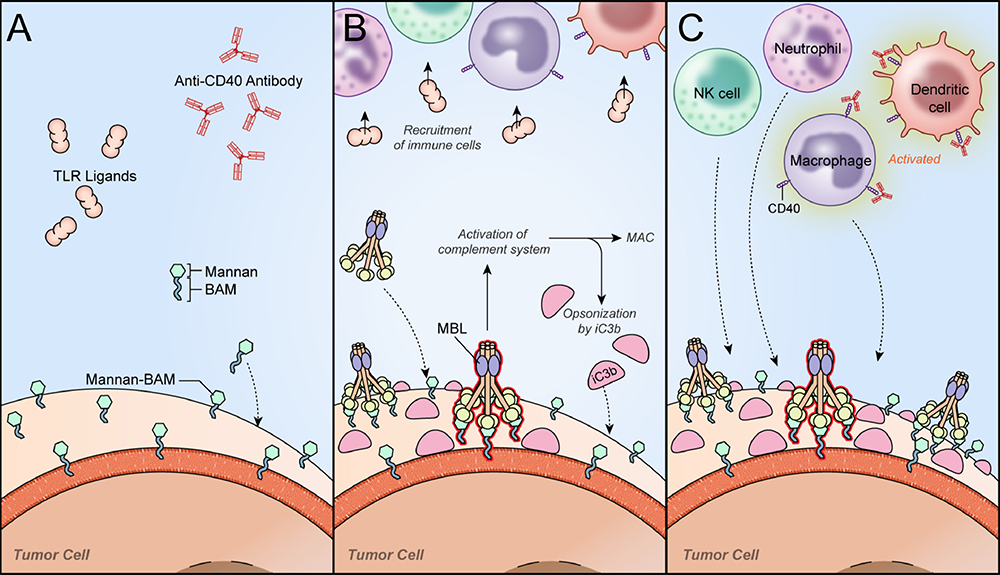
Click image to view.
The initial phase of immunotherapy based on the combination of TLR ligands, mannan–BAM, and anti–CD40 antibody.
A. After intra-tumoral application of the therapy (TLR ligands, mannan–BAM, anti–CD40 antibody), tumor cells are artificially opsonized by mannan–BAM, where the terminal part of BAM is incorporated into the lipid bilayer of tumor cells.
B. Mannan–BAM is subsequently recognized by mannan-binding lectin (MBL). The recognition results in activation of the complement system followed by proteolytic cleavage of complement protein C3 and the production of the terminal membrane attack complex (MAC). During the proteolytic cleavage of C3 into C3a and C3b, the inactive form of C3b (iC3b) opsonizes the tumor cells. Simultaneously, the TLR ligands R-848, poly(I:C), and LTA are recruiting immune cells into the tumor.
C. Tumor cells opsonized by iC3b are recognized by innate immune cells (NK cells, neutrophils, macrophages) previously recruited into the tumor. Such innate immune cells use their effector mechanisms to kill the opsonized tumor cells. As a part of the therapy, anti–CD40 antibodies bind to CD40 receptors expressed mainly on antigen-presenting cells (macrophages, dendritic cells) and initiate their activation. The activated antigen-presenting cells further internalize tumor antigens and present them to T cells in lymph nodes.
Therapeutic aspects of pheochromocytoma and paraganglioma
SDHB–mutated PPGLs exhibit dysregulation in oxygen-metabolic pathways, including pseudohypoxia and the formation of reactive oxygen species, suggesting that targeting the redox balance pathway is a potential therapeutic approach. By investigating PPGL cells with low SDHB levels, we showed that pseudohypoxia resulted in elevated expression of iron-transport proteins, including transferrin (TF), transferrin receptor 2 (TFR2), and the divalent metal transporter 1 (SLC11A2; DMT1), leading to iron accumulation. The iron overload contributed to elevated oxidative stress. At pharmacologic concentrations, ascorbic acid disrupted redox homeostasis, inducing DNA oxidative damage and cell apoptosis in PPGL cells with low SDHB levels. Moreover, using a preclinical animal model with PPGL allografts, we demonstrated that pharmacologic ascorbic acid suppressed SDHB–low metastatic lesions and prolonged overall survival. The data demonstrate that targeting redox homeostasis as a cancer vulnerability with pharmacologic ascorbic acid is a promising therapeutic strategy for SDHB–mutated PPGLs.
Mechanistically, nuclear factor erythroid 2–related factor 2 (NRF2)–guided glutathione de novo synthesis plays a key role in supporting cellular survival and the proliferation of SDHB–knockdown cells. We found that NRF2 blockade not only disrupted reactive oxygen species homeostasis in SDHB–deficient cells but also caused severe cytotoxicity by the accumulation of DNA oxidative damage. Brusatol, a potent NRF2 inhibitor, showed a promising effect in suppressing SDHB–gene metastatic lesions in vivo, with prolonged overall survival in mice bearing PPGL allografts. Our findings highlight a novel therapeutic strategy of targeting the NRF2–driven glutathione metabolic pathway against SDHB–mutated PPGLs.
PPGLs, arising from chromaffin cells, produce the catecholamines epinephrine and norepinephrine. The tumor biochemical phenotype is defined by which of these exerts the greatest influence on the cardiovascular system when released into circulation in high amounts. Action on the heart and vasculature can cause potentially lethal arrhythmias, often in the setting of comorbid blood pressure derangements. In a review of electrocardiograms obtained from PPGL patients (n = 650) treated at our institution over the last decade, we found severe and refractory sinus tachycardia, atrial fibrillation, and ventricular tachycardia to be the most common or life-threatening catecholamine-induced tachy-arrhythmias. These arrhythmias, arising from catecholamine excess rather than from a primary electro-physiologic substrate, require special considerations for treatment and complication avoidance. Understanding the synthesis and release of catecholamines, the adrenoceptors that catecholamines bind to, and the cardiac and vascular response to epinephrine and norepinephrine underlies optimal management in catecholamine-induced tachy-arrhythmias. Therefore, in a recent review we outlined tachy-arrhythmias in PPGLs and their treatment options.
As the member of the Working group on Endocrine Hypertension of the European Society of Hypertension, we outlined the newest approaches to the evaluation and treatment of a patient with PPGLs based on current knowledge in PPGL epidemiology, genetics, diagnosis, and treatment. We also wrote a review as a guide for practicing clinicians summarizing current management of PHEO/PGL according to tumor size, location, age of first diagnosis, presence of metastases, and especially underlying mutations, in the era of precision medicine.
Animal model of pheochromocytoma and cell culture studies
We previously identified the syndrome of multiple paragangliomas and pheochromocytomas, duodenal somatostatinoma, and polycythemia resulting from post-zygotic EPAS1 (HIF2A)-gain-of-function mutations (also called Pacak-Zhuang syndrome). The mutations, located in the oxygen-degradation domain (ODD) of hypoxia-inducible factor-2a (HIF-2a), have been shown to impair hydroxylation by prolyl hydroxylase domain–containing protein 2 (PHD2) and subsequent association with the von Hippel-Lindau (VHL) protein. In such a situation, degradation of HIF-2a is impaired, resulting in its stabilization, prolonged activation, lack of response to normal or increasing oxygen tension, and activation of the transcription of many genes participating in tumorigenesis. Recently, together with the collaboration of NCI investigators, we developed transgenic mice with a gain-of-function Epas1A529V mutation (corresponding to human EPAS1A530V), which demonstrated elevated levels of erythropoietin and polycythemia, a decreased urinary metanephrine-to-normetanephrine ratio, and increased expression of somatostatin in the ampullary region of duodenum. The findings demonstrate the vital roles of EPAS1 mutations in the syndrome development and the great potential of the Epas1A529V animal model for further pathogenesis and therapeutics studies. The model is also being used to study other malformations in animals as well as to match them with those seen in our patients (neurological, vascular, and ocular malformations).
Publications
- Malaza G, Brofferio A, Lin F, Pacak K. Ivabradine in catecholamine-induced tachycardia in a patient with paraganglioma. New Engl J Med 2019;380:1284–1286.
- Liu Y, Pang Y, Zhu B, Uher O, Caisova V, Huynh T, Taieb D, Hadrava Vanova K, Ghayee H, Neuzil J, Levine M, Yang C, Pacak K. Therapeutic targeting SDHB-mutated pheochromocytoma/paraganglioma with pharmacologic ascorbic acid. Clin Cancer Res 2020;26:3868–3880.
- Jochmanova I, Abcede A, Guerrero R, Malong C, Wesley R, Huynh T, Gonzales M, Wolf K, Jha A, Knue M, Prodanov T, Nilubol N, Mercado-Asis L, Stratakis C, Pacak. Clinical characteristics and outcomes of SDHB-related pheochromocytoma and paraganglioma in children and adolescents. J Cancer Res Clin Oncol 2020;146:1051–1063.
- Tella S, Jha A, Taieb D, Horvath K, Pacak K. A comprehensive review of evaluation and management of cardiac paragangliomas. J Am Coll Cardiol 2020;76:451–464.
- Gubbi S, Nazari M, Taieb D, Klubo-Gwiezdzinska J, Pacak K. Catecholamine physiology and its implications in COVID-19. Lancet Diabetes Endocrinol 2020;8(12):978–986.
Collaborators
- Zahraa Abdul Sater, MD, MPH, Clinical Endocrine Section, NIDDK, Bethesda, MD
- James Bibb, PhD, University of Alabama Comprehensive Cancer Center, University of Alabama at Birmingham Medical Center, Birmingham, AL
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Marilo Chiara, PhD, Hospital Universitario Central de Asturias, Oviedo, Spain
- Peter Deen, PhD, Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands
- Jaydira Del Rivero, MD, Pediatric Oncology Branch, NCI, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Stephanie Fliedner, PhD, Universitätsklinikum Schleswig-Holstein, Lübeck Medizinische Klinik I, Lübeck, Germany
- Zdenek Fryšák, MD, PhD, University Hospital and Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czech Republic
- Hans Ghayee, DO, Department of Internal Medicine, University of Florida, Gainesville, FL
- Peter Herscovitch, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Frank I. Lin, MD, Molecular Imaging Program, NCI, Bethesda, MD
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Renato Mariani-Constantini, MD, PhD, Aging Research Center (CeSI), D’Annunzio University, Chieti-Pescara, Italy
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Jirí Neužil, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Seyedehmoozhan Nikpanah, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda. MD
- Naris Nilubol, MD, FACS, Surgical Oncology Program, NCI, Bethesda, MD
- Rebecca Oakey, DPhil, King’s College, London, United Kingdom
- Ondrej Petrak, MD, PhD, Third Department of Medicine, General University Hospital, Prague, Czech Republic
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Mercedes Robledo, PhD, Human Cancer Genetics Programme, Spanish National Cancer Centre (CNIO), Madrid, Spain
- Jared Rosenblum, MD, NCI, NIH, Bethesda, MD
- Douglas Rosing, MD, Translational Medicine Branch, NHLBI, Bethesda, MD
- Kelly Roszko, MD, PhD, Skeletal Disorders and Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Babak Saboury, MD, Nuclear Medicine Department, CC, NIH, Bethesda MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Richard Tothill, PhD, University of Melbourne Centre for Cancer Research, Melbourne, Australia
- Victor Velculescu, MD, PhD, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD
- Brad Wood, MD, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Chunzhang Yang, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Deena Zeltser, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit http://pheopara.nichd.nih.gov.