Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Sakshi Chauhan, PhD, Visiting Fellow
- Lili Chen, PhD, Visiting Fellow
- Vishal Nehru, PhD, Visiting Fellow
- Keita Saeki, MD, PhD, Visiting Fellow
- Stella Zhu, BS, Postbaccalaureate Fellow
The laboratory is interested in chromatin and gene regulation in innate immunity. We study the role of the three nuclear factors histone H3.3, BRD4, and IRF8. Histone H3.3 is a variant H3 that is incorporated into nucleosomes along with transcriptional elongation, an unusual but defining feature of H3.3, given that most of other histones are deposited into nucleosomes during replication. For this reason, H3.3 is thought to be involved in epigenetic memory created by transcription, although experimental evidence for memory formation/maintenance is scant. BRD4 is a bromodomain protein of the BET family, expressed broadly in many cells, from early embryos to adults. Through the bromodomain, BRD4 binds to acetylated histones but not to unacetylated histones. BRD4 is thus called a “chromatin reader,” a type of regulatory factor capable of conveying epigenome information to other regulators of gene expression. Furthermore, BRD4, binds to the elongation factor complex P-TEFb through the C-terminal domain, and drives transcription of many genes by helping RNA polymerase II move through the gene body and to generate nascent mRNA. Many recent reports point out that BRD4 promotes growth of cancer cells, including various blood cancers, by mediating formation of super-enhancers. IRF8, which we reported in 1990 for the first time, is a DNA–binding transcription factor that plays an essential role in innate immune responses. IRF8 is expressed mostly in cells of the myeloid lineage, including monocytes/macrophages, dendritic cells, and microglia. IRF8 is strongly induced when stimulated by interferons (IFN). In addition, it is upregulated when myeloid cells encounter pathogen-derived molecules and those produced by stress. In turn, IRF8 activates many genes important for host resistance against pathogens. IRF8–induced genes include those involved in autophagy and lysosome-mediated pathogen clearance. IRF8 does so by binding to the promoter and enhancer regions of the target genes.
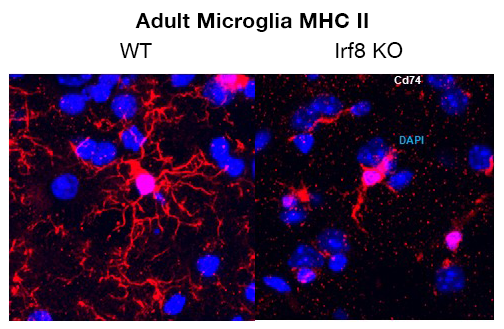
IRF8 shapes adult microglia identity through binding to super-enhancers.
IRF8, along with the transcription factor Spi1 (PU.1), plays a role in early embryonic development of microglia after the progenitors in the yolk sac migrate into the embryonic brain. It is assumed that IRF8 also has a role in adult microglia, given that it is expressed at high levels in such cells. However, the role of IRF8 in adult microglia has not been fully understood. Microglia from Irf8 knockout (KO) mice exhibit abnormal morphology and do not express MHC II on the cell surface (Figure 1). We are in the process of studying transcriptome profiles and epigenome landscapes of microglia with and without IRF8, using microglia purified from adult brain perfused from wild-type (WT) and Irf8 KO mice. Bulk and single-cell (sc) RNA-seq analyses found that, without IRF8, many genes that are defined as microglia identity were strongly downregulated, including microglia-specific cell-surface marker genes such as P2ry12, Iba1, Cx3cr1, and Ccr5. On the other hand, some of IFN–stimulated genes (ISGs) and disease-associated microglia (DAM) genes were aberrantly expressed in Irf8 KO microglia. In addition and most significantly, we found that IRF8 is required to activate two transcription factors critical for adult microglia identity and function, i.e., Sall1 and Batf3.
Figure 1.
Click image to view.
IRF8 is highly expressed in adult microglia and confers microglia cell identity. IRF8 KO microglia lack ramified extensions and fail to express MHC II (CD74).
Furthermore, we determined genome-wide IRF8 distribution in adult microglia using the CUT&RUN technique (which identifies the binding sites of DNA–associated proteins). Our analyses revealed that IRF8 binds mostly over distant enhancer regions, located upstream and downstream of its target genes; there was less IRF8 binding on the immediate promoter regions. Some of IRF8–bound sites were within the large “super-enhancers” enriched with H3K27ac histone marks, which are responsible for strong transcription of genes that create specific cell types. Irf8 KO microglia were devoid of super-enhancers important for shaping microglia identity. In accordance, an ATAC-seq (assay for transposase-accessible chromatin using sequencing) analysis found that IRF8 is important for installing open chromatin necessary for microglia super-enhancers and gene expression. The results imply that IRF8 critically contributes the formation of the microglia-specific epigenome landscape. Further analyses to delineate DNA methylome profiles are ongoing. Our analyses indicate that IRF8 regulates overall CpG–island methylation patterns. In a recently published study on a mouse model of Alzheimer’s disease (AD), it was suggested that IRF8 is involved in the progression of the disease. We investigated an AD mouse model (5FAD) with and without the Irf8 gene. Our transcriptome and other analyses indicate that IRF8 has dual roles and can promote AD pathogenesis in a complex manner.
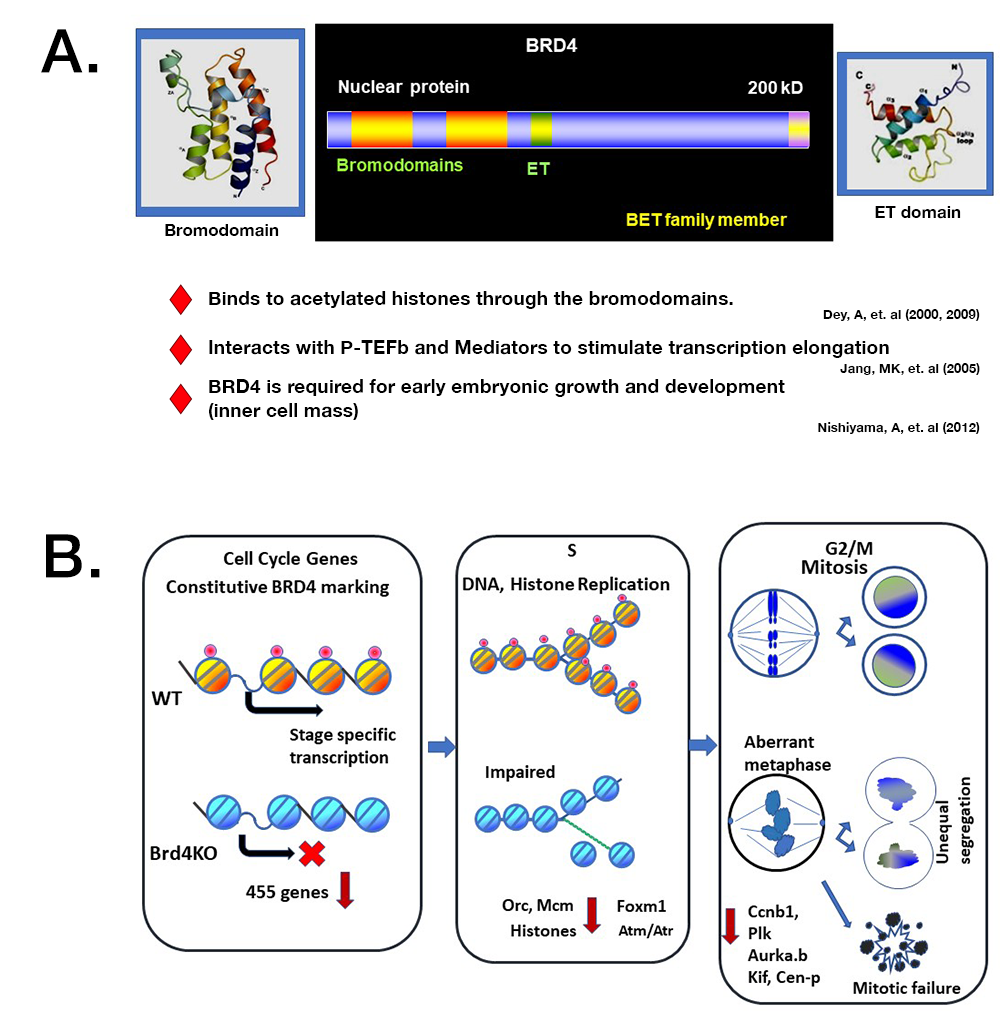
BRD4 marks cell-cycle genes and orchestrates gene expression necessary for mitotic cell division.
Cell-cycle progression is driven by sequential activation of transcription factors, kinases, and other effectors. BRD4 has been shown to promote the growth of many cancers. Recent publications report that several BRD4 inhibitors (BETi) can arrest cancer growth. BETi thus offer a new possibility for anti-cancer therapy. However, the role of BRD4 in controlling the growth of normal cells has remained elusive. We investigated whether BRD4 also regulates the growth of normal cells by examining embryonic fibroblasts from Brd4 conditional knockout (KO) mice. By conducting growth-curve analysis and an analysis the cell-cycle progression of synchronized cells, we found that BRD4 is critically required for cell-cycle progression of such cells from cell-cycle phases G0/G1, G1 to S and to G2/M. Transcriptome analysis found that genes required for every cell-cycle stage were markedly downregulated in Brd4 KO cells, including several histone genes at S phase, as well as the G2/M master regulators FOXM1 and ATM/ATR. FOXM1 is a transcription factor of the forkhead family and promotes transcription of many G2/M genes. ATM/ATR are kinases previously known to be involved in DNA–damage repair. ATR/ATM were recently shown to play a role in G2/M transition of normal cells. Consistent with these results, BRD4 constitutively occupied numerous cell-cycle genes active at cell-cycle phases G0/G1, S, and G2/M. Interestingly, overall BRD4 occupancy was maximum at S phase. ChIP-seq analysis of BRD4 binding revealed that BRD4 occupies numerous cell-cycle genes at the transcription start site (TSS) and gene body throughout the entire stages of growth. Thus cell-cycle genes are constitutively marked by BRD, which would provide a platform for continuous cell proliferation. We performed time-lapse imaging analysis to determine the consequence of BRD4 deficiency. We found that the absence of BRD4 culminates in G2/M cells undergoing a catastrophic failure in mitosis. Nuclear materials in Brd4 KO cells disintegrated without assembling at the mitotic plate, leading to apoptosis (Figure 2B). Our study demonstrates that BRD4 orchestrates the entire program of cell-cycle progression for both normal and cancer cells.
Figure 2.
Click image to view.
A. BRD4 has two bromodomains and an ET domain. The lab characterized essential features of this factor.
B. Model for BRD4 regulation of cell cycle progression
Top. In normal cells (WT), BRD4 binds to cell-cycle genes and drives their transcription in a stage-specific manner and orchestrates mitotic cell division.
Bottom. In Brd4 KO cells, cell-cycle genes are not marked by BRD4, and their expression is down-regulated and delayed. Brd4 KO cells are defective in chromatin duplication at S phase and fail to execute mitotic cells division. Many Brd4 KO cells undergo apoptotic cell death, while some KO produce offspring with unequal chromosomal segregation.
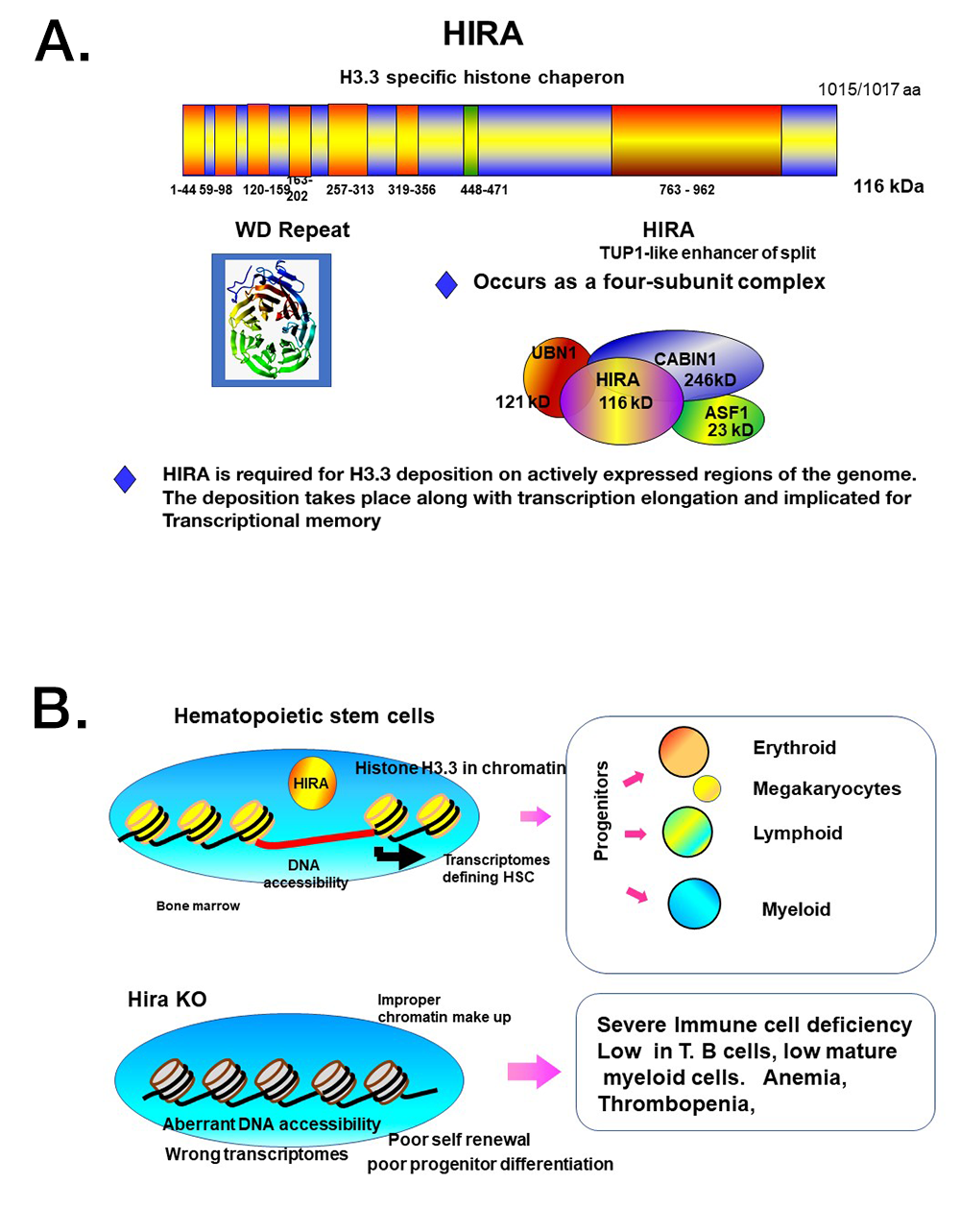
HIRA confers chromatin accessibility on the hematopoietic stem cells and guides entire hematopoietic lineage development.
We have been interested in the role of histone H3.3, given that it is implicated in transcriptional memory. H3.3 has been shown to localize to actively transcribed and bivalent regions, a process mediated by the H3.3–specific histone chaperone HIRA. H3.3 also localizes to heterochromatin regions including the telomere, where transcription is silenced. The latter process is mediated by another chaperone, ATRX. With the aim of developing experimental tools useful for clarifying localization and the functional significance, we constructed firstly mouse lines expressing HA–tagged H3.3 and secondly those with conditional H3.3 knockout. Both H3.3 genes (H3f3a and H3f3b) were replaced by H3.3–HA and marked by LoxP (sites that denote nucleotide sequences removable by Cre recombinase). By studying the H3.3–HA mice, we showed that H3.3 is expressed in almost all adult tissues, including immune cells. Genome-wide deposition analysis of embryonic fibroblasts revealed that H3.3–HA is deposited over numerous genes, approximately correlating with levels of mRNA expression.
We are also interested in studying the role of HIRA, because it is involved in genic H3.3 deposition and is likely to control transcription programs and memory formation (Figure 3B). We therefore constructed Hiraf/f mice and generated mice in which the Hira gene is deleted in hematopoietic stem cells (HSCs) by crossing Hiraf/f with the Vav-Cre strain. The mice provide a novel tool with which to elucidate the role of HIRA in hematopoiesis, which is not well understood. The study has some clinical relevance in that patients with DiGeorge syndromes have deletion of an over 11Mb DNA stretch encompassing HIRA. DiGeorge syndromes are little understood diseases, and patients manifest varied abnormalities, including immunodeficiency and thrombocytopenia. Our analysis found that HIRA is essential for the development and the maintenance of bone marrow (BM) HSCs, in that long-term (LT) HSC were almost absent in Hira KO BM. LT-HSCs possess the capacity for self-renewal and progenitor differentiation. Because all adult hematopoietic cells are derived from the HSCs, Hira KO mice were deficient in all three blood lineages, erythroid, myeloid, and lymphoid cells, leading to anemia, thrombocytopenia, and severe immunodeficiency. While some T cells were found in peripheral lymphoid organs, including thymus and lymph nodes, mature B cells were virtually absent from the mice. Such deficiencies were replicated in adoptive transfer experiments, in which wild-type mice reconstituted with Hira KO HSCs failed to generate erythroid, myeloid, and lymphoid lineage cells of Hira KO origin. However, fetal hematopoiesis was normal in Hira KO mice, although fetal HSCs lacked reconstitution capacity. Transcriptome analysis revealed that HIRA is required for expression of many transcription factors and signaling molecules critical for HSCs. ATAC-seq analysis revealed that HIRA establishes HSC–specific DNA accessibility, including the SPI1/PU.1 sites (see Figure 3B for a model). The study demonstrates that HIRA creates a chromatin environment essential for HSCs to acquire self-renewal and differentiation capacity.
Figure 3.
Click image to view.
A. The histone chaperone HIRA specifically deposits H3.3 on genic regions. It has a WD repeat domain and TUP1 (a transcriptional repressor), conserved from yeast to humans.
B. Model for the function of HIRA in HSCs
HSCs possess self-renewal and differentiation capacity. HIRA has a central role in conferring HSC–specific chromatin accessibility, necessary for HSC–specific transcriptome programs. In the absence of Hira, proper chromatin accessibility is not established, leading to aberrant transcription profiles. Phenotypically, the number of HSCs are dramatically reduced. Consequently, Hira KO mice present with severe immunodeficiency, anemia, and thrombopenia.
Additional Funding
- NICHD Scientific Director's Award (2018–2019)
- Washington University RVCL (Retinal Vasculopathy with Cerebral Leukodystrophy) Foundation
Publications
- Bachu M, Tamura T, Chen C, Narain A, Nehru V, Sarai N, Ghosh SB, Ghosh A, Kavarthapu R, Dufau ML, Ozato K. A versatile mouse model of epitope-tagged histone H3.3 to study epigenome dynamics. J Biol Chem 2019;294:1904-1914.
- Dey A, Yang W, Gegonne A, Nishiyama A, Pan R, Yagi R, Grinberg A, Finkelman FD, Pfeifer K, Zhu J, Singer D, Zhu J, Ozato K. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J 2019;38(7):e100293.
- Thumbigere-Math V, Foster BL, Bachu M, Yoshii H, Brooks SR, Coulter A, Chavez MB, Togi S, Neely AL, Deng Z, Mansky KC, Ozato K, Somerman MJ. Inactivating mutation in IRF8 promotes osteoclast transcriptional programs and increases susceptibility to tooth root resorption. J Bone Miner Res 2019;34(6):1155-1168.
- Chen C, Sun M-A, Warzecha C, Bachu M, Dey A, Wu T, Adams P, Macfarlan T, Love P, Ozato K. HIRA, a DiGeorge syndrome candidate gene, confers proper chromatin accessibility on HSCs and supports all stages of hematopoiesis. Cell Rep 2020;30(7):2136-2149.
- Miyagawa F, Tagaya Y, Ozato K, Horie, K, Asada H. Inflammatory monocyte-derived dendritic cells mediate autoimmunity in murine model of systemic lupus erythematosus. J Transl Autoimmun 2020;3:100060.
Collaborators
- Steven L. Coon, PhD, Molecular Genomics Core, NICHD, Bethesda, MD
- Robert J. Crouch, PhD, Section on Formation of RNA, NICHD, Bethesda, MD
- Haitong Hou, PhD, Laboratory of Biochemistry and Molecular Biology, National Cancer Institute, Bethesda, MD
- Yoh-suke Mukouyama, PhD, Laboratory of Stem Cell and Neuro-Vascular Biology, NHLBI, Bethesda, MD
- Dinah S. Singer, PhD, Experimental Immunology Branch, NCI, Bethesda, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
- Vivek Thumbigere Math, DDS, PhD, University of Maryland, School of Dentistry, Baltimore, MD
Contact
For more information, email ozatok@mail.nih.gov or visit http://ozatolab.nichd.nih.gov.