Chromatin Remodeling and Gene Activation

- David J. Clark, PhD, Head, Section on Chromatin and Gene Expression
- Peter Eriksson, PhD, Staff Scientist
- Christopher Coey, PhD, Intramural Research Training Award Fellow
- Hemant K. Prajapati, PhD, Visiting Fellow
- Allison F. Dennis, PhD, Intramural Research Training Award Fellow
- Paul Elizalde, BS, Postbaccalaureate Fellow
- LauraAnn Schmidberger, BS, Postbaccalaureate Fellow
Aberrant gene regulation is the basis of many disease states. Our main objective is to understand how genes are activated for transcription in the context of chromatin structure. Chromatin is not merely a packaging system for DNA in eukaryotic cells; it also participates in gene regulation. The structural subunit of chromatin is the nucleosome, which contains nearly two turns of DNA coiled around a central core histone octamer. Nucleosomes are generally quite regularly spaced along the DNA, like beads on a string. Gene regulation involves either attenuation of the inherently repressive properties of nucleosomes to facilitate gene expression, or enhancement of those properties to ensure complete repression, events that are choreographed by DNA sequence–specific transcription factors (activators and repressors) and chromatin-modifying complexes. The latter can be divided into two groups: histone- or DNA–modifying enzymes that implement the “epigenetic code”, and ATP–dependent remodeling machines that move or displace nucleosomes. We are exploiting and developing high-throughput technologies to obtain genome-wide maps of nucleosomes, chromatin-remodeling complexes and RNA polymerase II in budding yeast to determine how chromatin organization is affected when genes are activated. The current objectives of our yeast studies are: (1) to determine the roles of the various chromatin-remodeling complexes (RSC, SWI/SNF, ISW1, ISW2, CHD1, Ino80C) in chromatin organization and gene expression, why there are so many different remodelers, and whether they are functionally redundant; our studies so far indicate that each remodeling enzyme makes a different contribution to chromatin organization; and (2) to test the hypothesis that nucleosomes control DNA accessibility and play a vital role in gene regulation by blocking promoters. We are also extending our studies of chromatin remodeling from yeast to higher organisms.
Many human diseases have been linked to chromatin remodeling enzymes and epigenetic modifications. For example, mutations in the gene encoding the hSNF5 subunit of the SWI/SNF ATP–dependent chromatin-remodeling complex are strongly linked to pediatric rhabdoid tumors. The CHD class of ATP–dependent remodelers has also been linked to cancer and autism. Cancer therapies and drugs aimed at epigenetic targets are being tested. A full understanding of chromatin structure and the mechanisms by which it is manipulated is therefore of vital importance.
Chromatin remodeling and DNA accessibility
Gene activation involves the recruitment of a set of factors to a promoter in response to appropriate signals, ultimately resulting in the formation of an initiation complex by RNA polymerase II and transcription, events that coincide with the removal of promoter nucleosomes to create a nucleosome-depleted region (NDR). This observation led to the generally accepted model that promoter nucleosomes physically block transcript initiation, acting as repressors by preventing access to specific transcription factor binding sites. The nucleosome is a highly stable structure containing tightly wound DNA, which is largely inaccessible to sequence-specific DNA–binding proteins. Activation occurs if sequence-specific ‘pioneer’ transcription factors are present (proteins that bind to nucleosomal sites with high affinity), and/or if ‘classical’ transcription factors, which are normally blocked by nucleosomes, recruit ATP–dependent chromatin remodelers to move or evict promoter nucleosomes, thus facilitating initiation complex formation.
The ATP–dependent chromatin remodelers variously move nucleosomes along DNA, remove the histones altogether, or form arrays of regularly spaced nucleosomes. Examples include the SWI/SNF and RSC complexes, which remodel nucleosomes on genes and at promoters, and the CHD and ISWI complexes, which are often involved in determining nucleosome spacing. The Ino80C complex is unusual because it has both properties. We wrote a review describing the ATP–dependent remodelers and their roles in chromatin organization [Reference 1].
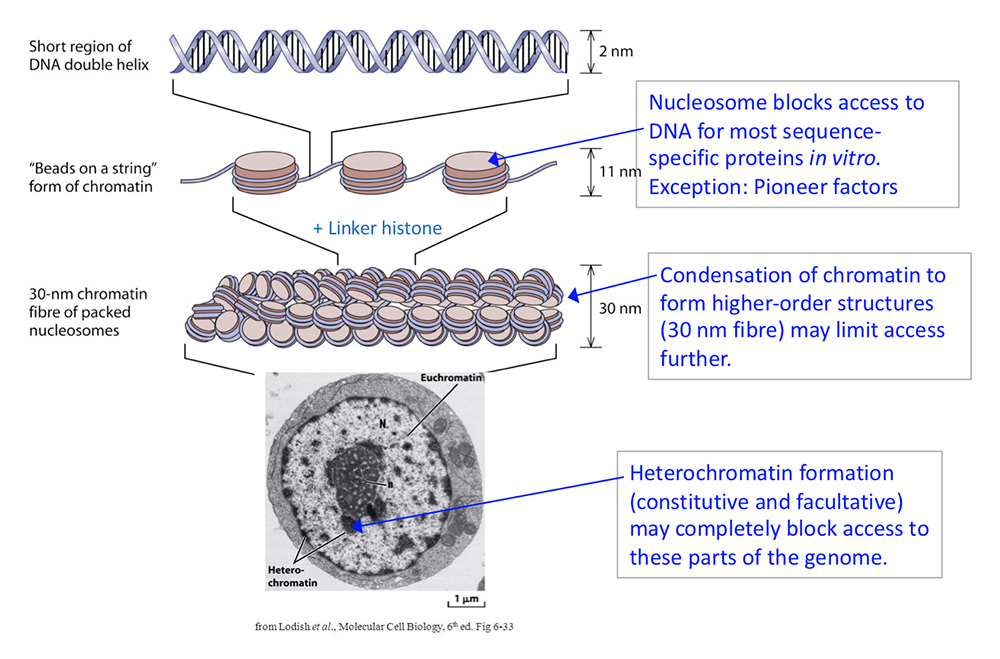
Figure 1. DNA packaging in the nucleus: to what extent does chromatin compaction limit access to the DNA?
Click image to view.
DNA is packaged into the nucleus by histones. The basic structural subunit of chromatin is the nucleosome core, which contains about 147 bp of DNA wrapped nearly twice around a central octamer of core histones. Nucleosomes are regularly spaced along the DNA like beads on a string; the intervening DNA is called the linker DNA and is bound by linker histone (H1). The beads-on-a-string fiber spontaneously condenses into a heterogeneous fiber of about 30 nm width. Genomic regions rich in repetitive elements form constitutive heterochromatin in all cells, in which the chromatin fiber is even more condensed. Facultative heterochromatin is formed on genes that should be permanently silent in a specific differentiated cell type. Heterochromatin is densely packed and darkly staining in the electron micrograph shown here. Euchromatin is less condensed (light staining) and contains active genes. We are interested in determining to what extent chromatin limits DNA accessibility. Figure adapted from Chereji et al. Genome Res 2019;29:1985-1995.
Chromatin may block access to the DNA at several levels: the nucleosome itself, the higher order coiling of the nucleosomal fiber, and further condensation of nucleosomal fibers into heterochromatin (Figure 1). We have begun to test the hypothesis that DNA accessibility is of major importance in regulating gene expression. To do this, we needed a fully quantitative measure of DNA accessibility. Accordingly, we used the restriction enzyme AluI as a probe of chromatin structure and as a proxy for transcription factors, given that both bind to specific DNA sequences. We measured the digestion rate and the fraction of accessible DNA accurately at all genomic AluI sites in budding yeast nuclei and in mouse liver-cell nuclei. We observed that mouse liver DNA is significantly more accessible than yeast DNA, which can be explained simply by recognizing that the linker DNA between nucleosomes is much more accessible than nucleosomal DNA and that nucleosomes are spaced farther apart in mouse chromatin, resulting in longer linkers than in yeast chromatin, data indicating that nucleosome spacing is a major determinant of accessibility. More remarkable is the fact that DNA accessibility is binary, such that each site is accessible in some cells (i.e., in a linker and cut by AluI) and essentially completely inaccessible in the remaining cells (i.e., nucleosomal and resistant to AluI). In fact, we found no sites that are accessible in every cell or inaccessible in every cell, an observation that indicates that nucleosome positioning is generally imperfect, even at promoters, such that nucleosomes never reliably block a specific site. For example, AluI sites in inactive mouse promoters are accessible in some cells, even though the gene is inactive, implying that the nucleosome is insufficient to block transcription factor binding in all cells and suggesting that the simple promoter nucleosome block model is incorrect (Figure 2). Surprisingly, in mouse nuclei, the relatively decondensed euchromatin (which contains mostly active genes) and the highly condensed heterochromatin (which contains mostly inactive genes) have very similar accessibilities to AluI, suggesting that transcription factors could also penetrate heterochromatin. Overall, our observations suggest that DNA accessibility is not likely to be the primary determinant of gene regulation.
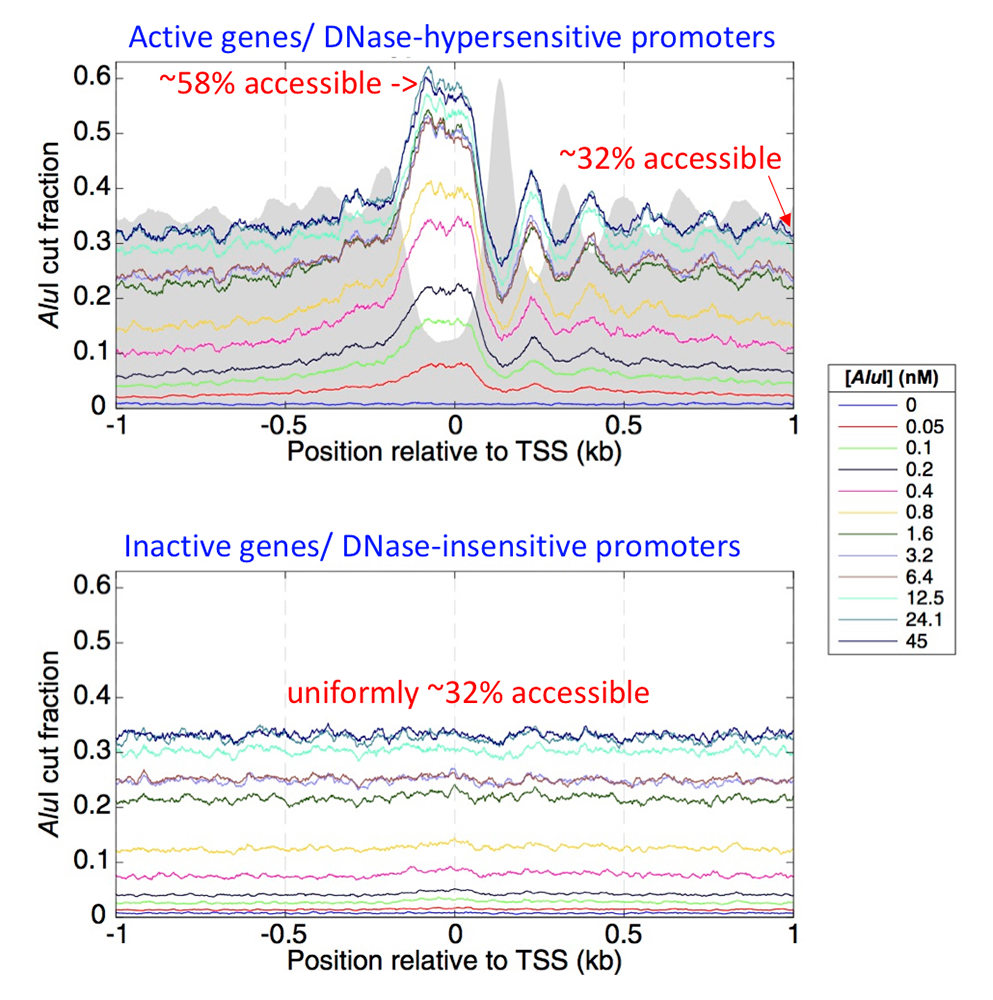
Figure 2. Accessibility of active and inactive promoter DNA in mouse liver cells
Click image to view.
Mouse genes were divided into active and inactive genes, defined by the hypersensitivity of their promoters to DNase I, which correlates with transcription levels. Active genes have nucleosome-depleted promoters, which are more sensitive to digestion by DNase I. The promoters of such genes are aligned on their transcription start sites (TSS). The grey background indicates average nucleosome positions from MNase-seq data for the same cells. The fraction of the DNA accessible to the AluI restriction enzyme was calculated as a function of AluI concentration. The fraction cut by AluI reaches a limit value of about 32% at higher AluI concentrations in promoter-proximal regions, except at the nucleosome-depleted promoters of active genes, where it reaches a limit value of about 58%. For active genes, the fraction cut by AluI is higher in the linkers than inside nucleosomes (the AluI and nucleosome peaks are out of phase). Inactive gene promoters are not nucleosome-depleted and show no nucleosome phasing. However, they are accessible in about 32% of the cells, even though they are inactive. Figure adapted from Chereji et al. Genome Res 2019;29:1985-1995.
Differential nucleosome spacing in neurons and glia
Most eukaryotic cells have a characteristic average nucleosome spacing of about 190 bp, corresponding to a linker of about 45 bp. However, cortical neurons have a shorter average spacing of 165 bp, similar to that of the yeasts. The significance of this atypical global chromatin organization is unclear. In a collaboration with Doug Fields [Reference 2], we compared the chromatin structures of purified mouse dorsal root ganglia (DRG) neurons, cortical oligodendrocyte precursor cells (OPCs), and cortical astrocytes. We found that DRG neurons have short average spacing (165 bp), whereas OPCs (182 bp) and astrocytes (183 bp) have longer spacing. Most genes in all three cell types have a promoter chromatin organization typical of active genes: a promoter NDR flanked by regularly spaced nucleosomes. In DRG neurons, nucleosome spacing downstream of promoters is longer than expected from the genomic average, whereas nucleosome spacing in OPCs and astrocytes is similar to the global average for these cells. Thus, the atypical nucleosome spacing of neuronal chromatin does not extend to promoter-proximal regions. Although we gained some insight into the role of nucleosome spacing in different cell types, its significance remains unclear.
Most eukaryotic cells have a characteristic average nucleosome spacing of about 190 bp, corresponding to a linker of about 45 bp. However, cortical neurons have a shorter average spacing of 165 bp, similar to that of the yeasts. The significance of this atypical global chromatin organization is unclear. In a collaboration with Doug Fields [Reference 2], we compared the chromatin structures of purified mouse dorsal root ganglia (DRG) neurons, cortical oligodendrocyte precursor cells (OPCs), and cortical astrocytes. We found that DRG neurons have short average spacing (165 bp), whereas OPCs (182 bp) and astrocytes (183 bp) have longer spacing. Most genes in all three cell types have a promoter chromatin organization typical of active genes: a promoter NDR flanked by regularly spaced nucleosomes. In DRG neurons, nucleosome spacing downstream of promoters is longer than expected from the genomic average, whereas nucleosome spacing in OPCs and astrocytes is similar to the global average for these cells. Thus, the atypical nucleosome spacing of neuronal chromatin does not extend to promoter-proximal regions. Although we gained some insight into the role of nucleosome spacing in different cell types, its significance remains unclear.
A method for assessing histone surface accessibility genome-wide
Almost all genomic methods focus on the DNA in chromatin. We have begun to study how histone accessibility might be altered in the cell in response to transcription. For example, there is some evidence in the literature that nucleosomes can undergo a major conformational change, exposing internal surfaces of the histone octamer. In a collaboration with Jeffrey Hayes, we developed a method to assess exposure of histone protein surfaces in yeast chromatin [Reference 3]. A histone amino acid residue on the octamer surface (or buried within the octamer) is substituted with cysteine, and its exposure is measured by reaction with a thiol-specific reagent. Nuclei are treated with biotin-maleimide and then the chromatin is digested to nucleosomes by micrococcal nuclease. Biotinylated nucleosomes are purified using streptavidin beads. Nucleosomal DNA from input and affinity-purified samples are sequenced and compared. Currently, we are assessing the exposure of several different histone surfaces within active and inactive chromatin.
Role of the Ino80C chromatin remodeler at highly expressed genes in yeast
We continued our collaboration with Alan Hinnebusch, publishing several papers together concerning the roles of the SWI/SNF and RSC chromatin remodelers in promoter nucleosome eviction. We have now studied the role of the Ino80 remodeling complex (Ino80C) in transcriptional activation [Reference 4]. We showed that Ino80C is important for promoter nucleosome eviction and transcriptional activation. Compared with SWI/SNF, Ino80C generally functions over a wider region, spanning the promoter-flanking nucleosomes and NDR, at genes highly dependent on its function. Nucleosome eviction defects in cells lacking the Ino80 ATPase subunit are frequently accompanied by reduced promoter occupancies of TATA–binding protein (TBP), and diminished transcription. We concluded that Ino80C acts widely in the yeast genome, together with RSC and SWI/SNF, to evict promoter nucleosomes and enhance transcription.
Mechanisms preventing mislocalization of centromeric histone H3
Correct localization of the centromeric histone H3 variant (Cse4 in yeast; CENP-A in humans) to centromeres is vital for faithful chromosome segregation. Over-expression and mislocalization of CENP-A has been observed in many cancers and correlates with increased invasiveness and poor prognosis. In a collaboration with Munira Basrai's lab, we investigated the mechanism by which yeast cells prevent mislocalization of centromeric H3. The Basrai Lab identified yeast genes required to prevent Cse4 mislocalization using a genetic screen [Reference 5]. They include two F-box proteins, Met30 and Cdc4, which interact and cooperatively regulate Cse4 proteolysis, preventing its incorporation into non-centromeric nucleosomes and consequent chromosome instability. The Dbf4-dependent kinase complex also plays a role in Cse4 proteolysis, probably through the Psh1 E3 ubiquitin ligase [Reference 6].
Publications
- Prajapati HK, Ocampo J, Clark DJ. Interplay among ATP-dependent chromatin remodelers determines chromatin organisation in yeast. Biology 2020;9:190.
- Clark SC, Chereji RV, Lee PR, Fields RD, Clark DJ. Differential nucleosome spacing in neurons and glia. Neurosci Lett 2020;714:134559.
- Marr LT, Clark DJ, Hayes JJ. A method for assessing histone surface accessibility genome-wide. Methods 2019 184:61-69.
- Qiu H, Biernat E, Govind CK, Rawal Y, Chereji RV, Clark DJ, Hinnebusch AG. Chromatin remodeler Ino80C acts independently of H2A.Z to evict promoter nucleosomes and stimulate transcription of highly expressed genes in yeast. Nucleic Acids Res 2020;48:8408-8430.
- Au WC, Zhang T, Mishra PK, Eisenstatt JR, Walker RL, Ocampo J, Dawson A, Warren J, Costanzo M, Baryshnikova A, Flick K, Clark DJ, Meltzer PS, Baker RE, Myers C, Boone C, Kaiser P, Basrai MA. Skp, Cullin, F-box (SCF)-Met30 and SCF-Cdc4-mediated proteolysis of CENP-A prevents mislocalization of CENP-A for chromosomal stability in budding yeast. PLoS Genet 2020;16:e1008597.
- Eisenstatt JR, Boeckmann L, Au WC, Garcia V, Bursch L, Ocampo J, Costanzo M, Weinreich M, Sclafani RA, Baryshnikova A, Myers CL, Boone C, Clark DJ, Baker R, Basrai MA. Dbf4-dependent kinase (DDK)-mediated proteolysis of CENP-A prevents mislocalization of CENP-A in Saccharomyces cerevisiae. G3 (Bethesda) 2020;10:2057-2068.
Collaborators
- Munira A. Basrai, PhD, Genetics Branch, Center for Cancer Research, NCI, Bethesda, MD
- Harold Burgess, PhD, Section on Behavioral Neurogenetics, NICHD, Bethesda, MD
- Douglas Fields, PhD, Section on Nervous System Development and Plasticity, NICHD, Bethesda, MD
- Chhabi Govind, PhD, Oakland University, Rochester, MI
- Jeffrey J. Hayes, PhD, University of Rochester Medical Center, Rochester, NY
- Alan G. Hinnebusch, PhD, Section on Nutrient Control of Gene Expression, NICHD, Bethesda, MD
- Philip R. Lee, PhD, Section on Nervous System Development and Plasticity, NICHD, Bethesda, MD
- Vasily M. Studitsky, PhD, Fox Chase Cancer Center, Philadelphia, PA
Contact
For more information, email clarkda@mail.nih.gov or visit http://clarklab.nichd.nih.gov.