The Biological Impact of Transposable Elements

- Henry L. Levin, PhD, Head, Section on Eukaryotic Transposable Elements
- Angela Atwood-Moore, BA, Senior Research Assistant
- Paul Atkins, PhD, Postdoctoral Fellow
- Hyo Won Ahn, PhD, Visiting Fellow
- Feng Li, PhD, Visiting Fellow
- Rakesh Pathak, PhD, Visiting Fellow
- Angelique Ealy, BA, Postbaccalaureate Fellow
- Kyla Roland, BA, Postbaccalaureate Fellow
- Katie Wendover, BA, Postbaccalaureate Fellow
Mobile genetic elements create genetic diversity, which, through natural selection, promotes adaptation and innovation. A wealth of examples document how the action of transposable elements (TEs) leads to important modifications in morphology, metabolism, and ultimately fitness. Recent work reveals that large gene networks co-regulated by key transcription factors are built on regulatory sequences derived from TEs. Evolution of the networks depends on the dispersal of regulatory elements to genes that, when coexpressed, provide the host with novel functions such as innate immunity or the pregnancy of mammals. The adaptation and innovation produced by mobile elements comes at the cost of gene disruption resulting from indiscriminate insertion and homologous recombination. In each host species a tenuous balance is struck between the mutagenic activity of TEs and the benefits provided by the genetic diversity they produce, a genetic conflict that is affected by many factors, including host mechanisms that silence TEs and the activity of mobile elements, which can be triggered by environmental stress. When the balance between genome defense and TE activity is perturbed, transposition in gonads leads to infertility and, in the case of humans, de novo inserts generate disease alleles. The HIV-1 virus is an example of an infectious mobile element that can overwhelm defense mechanisms. Its efficient integration into the genome of immune cells has resulted in over 35 million deaths.
The long terminal repeat (LTR) retrotransposons of yeast provide a unique opportunity to study in real time the biology and impact of TEs in highly characterized model systems. The Ty1, Ty3, and Ty5 elements of Saccharomyces cerevisiae each possess unique mechanisms that limit the disruption of important sequences by directing integration to “safe havens” such as heterochromatin and sequences upstream of RNA polymerase III (pol III) promoters. Our studies on the LTR retrotransposon Tf1 of Schizosaccharomyces pombe revealed integration behavior that contrasts sharply with that of the Ty elements. Our large datasets of de novo integration show that Tf1 integrates principally into the promoters of RNA pol II–transcribed genes. Promoters are clearly not “safe havens,” raising questions about the biological significance of this integration behavior. Several experiments from our lab showed that Tf1 integration alters promoter activity and is well-suited to promote adaptation to environmental stress.
Transposable element insertions in fission yeast drive adaptation to environmental stress.
Cells are regularly challenged by environmental stress, to which rapid and robust responses are critical for survival. To cope with adverse conditions, cells activate transient programs of transcription that alter expression of hundreds to thousands of genes. Pre-wired transcription responses have evolved as the result of frequent exposure to a common set of external stresses. However, it is not clear how cells cope when confronted with environmental shock, defined here as novel stresses or conditions for which existing responses are inadequate to support survival. Although genetic modifications that improve survival can clearly be achieved through single-nucleotide mutations, these are mostly neutral or detrimental, and would rarely allow survival to abrupt changes in environment. Alternatively, TEs are activated by stress and, because they carry regulatory sequence, TEs can readily alter gene expression. Such a possibility is in line with McClintock’s original proposal that TEs provide a means to overcome the threat of environmental stress by reorganizing the genome [McClintock B. Science 1984;226:792]. However, the tenet has not been directly tested. Tf1 provides a unique opportunity to study the role of transposition in cells' survival in response to changes in environmental conditions. A stress-response enhancer embedded in Tf1 causes integration to induce the expression of adjacent promoters. The prominent clustering of integration in promoters and the influence of the Tf1 enhancer on adjacent genes suggest the intriguing possibility that Tf1 may be wired to provide efficient adaptation to environmental stress.
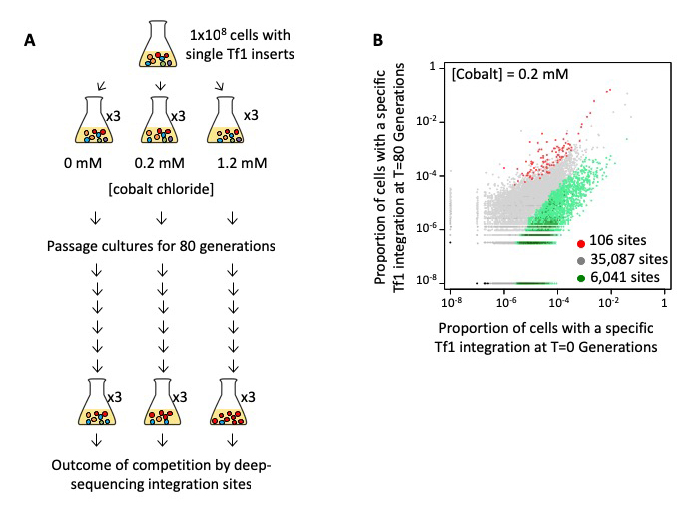
To test directly whether transposition plays a significant role in adaptation, we passaged populations of cells in medium containing various concentrations of CoCl2, which generates reactive oxygen species, causes DNA damage, induces apoptosis, and mimics hypoxia. We used cultures in which each cell contained one of 41,000 pre-established insertions of Tf1-neo, which we created by overexpression of Tf1 followed by selection for integration events (Figure 1A). Each cell contained a specific insertion that served as a tag, which we used to measure clonal expansion during competitive growth. With high-throughput sequencing of the Tf1-neo tags, we monitored clonal expansion in cultures grown for 80 generations in 0.0 mM, 0.2 mM, or 1.2 mM CoCl2. Three independent passaging experiments were conducted for each CoCl2 concentration. We assessed whether the Tf1 insertions themselves provided a prominent path to improved growth by identifying clones that expanded in each of the three independent passaging experiments. In cultures grown without CoCl2, the bulk of the insertions maintained constant proportions in the cultures. However, large changes occurred in the cultures that contained CoCl2. With 0.2 mM CoCl2, cells with 106 integration positions reproducibly expanded two-fold or more (Figure 1B). The positions accounted for 3.1% of the initial culture and expanded markedly to become 58%, 52%, and 31% (average of 47%) of the final cultures for passaging experiments 1, 2, and 3, respectively. Such substantial percentages of reproducible tags indicate that the Tf1 insertions contributed significantly to improved growth. Importantly, a significant proportion of the competition-enriched positions were adjacent to genes that participate in TOR pathways (signaling pathways that integrate both intracellular and extracellular signals and serve as a central regulators of cell metabolism, growth, proliferation, and survival), indicating that TOR provides resistance to CoCl2. In S. pombe, the TORC1 and TORC2 kinase pathways activate genes involved in cell growth and stress response, respectively. Deletion of genes involved in both TORC1 and TORC2 pathways, together with studies of strains with recreated insertions, demonstrated that Tf1 integration functioned as the major path to adaptation. To test whether TEs mediate adaptation in natural settings, we analyzed the genomes of 57 wild isolates of S. pombe. We found that polymorphic LTR insertions clustered significantly adjacent to genes that contribute to sporulation frequency, heat shock, and osmotic stress. Our analysis thus indicates that Tf elements function in the wild to provide resistance to environmental stress.
Figure 1. A reproducible set of Tf1 integration tags showed improved growth in CoCl2.
Click image to view.
A. Cells with Tf1 integration tags were passaged for 80 generations in CoCl2. Changes in clonal populations were monitored by sequencing the tags at T = 0 and T = 80 generations.
B. Proportions of cells in the cultures containing a Tf1 integration tag at each of the insertion sites at the beginning (T = 0; x-axis) versus the end (T = 80; y-axis) of the passages in the presence of 0.2 mM CoCl2.
Dense transposon integration reveals that essential cleavage and polyadenylation factors promote heterochromatin formation.
In eukaryotes, the assembly of DNA into highly condensed heterochromatin is critical for a broad range of functions related to genome integrity. The methylation of histone H3 on lysine 9 (H3K9me) is central to the formation of heterochromatin by creating binding sites for a range of chromatin proteins important for silencing transposable elements, chromosome segregation, and epigenetic inheritance. Used extensively for this purpose, S. pombe is an excellent model in which to study the molecular mechanisms that generate and regulate heterochromatin. Centromeres, subtelomeres, and the mating-type region are packaged into constitutive heterochromatin, while meiosis genes are silenced by facultative heterochromatin until cells are starved of nitrogen. Importantly, Clr4, the H3K9–specific histone methyltransferase, is recruited to heterochromatin regions by several mechanisms. Constitutive heterochromatin results from RNAi factors that include the Ago1–containing, RNA–induced transcriptional silencing complex (RITS). Facultative heterochromatin at meiosis genes is independent of RNAi and relies on the RNA elimination (i.e., degradation) factors Red1 and Mmi1 and on the nuclear exosome. However, gaps exist in our understanding of how RNA elimination generates heterochromatin. A new approach for identifying gene function is the high-throughput sequencing of integration profiles, also known as Tn-seq, which identifies genes important for growth under selective conditions. Genes necessary to sustain growth under a specific condition do not tolerate insertions in that condition. Tn-seq has been applied to identify pathogenic genes in bacteria. However, we were the first to develop the method for a eukaryote; we developed a method for identifying essential genes in yeast, and others have subsequently applied the strategy to single-cell eukaryotes.
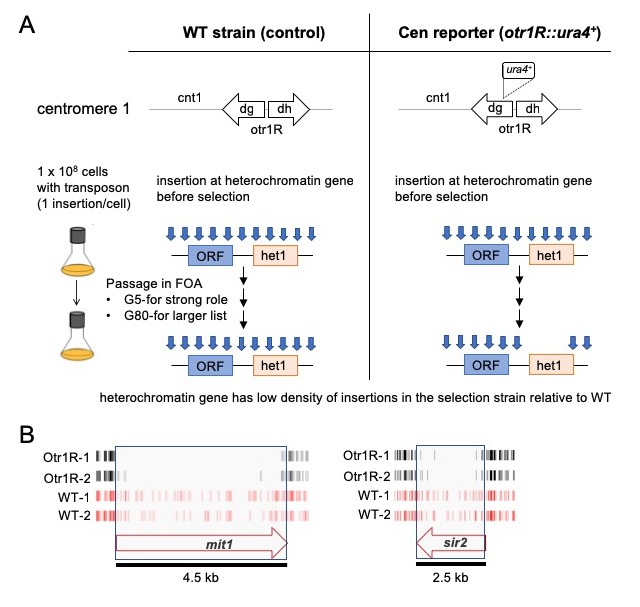
With the goal of identifying novel factors important for heterochromatin, we produced dense profiles of integrations using the Hermes transposable element and a silencing reporter (ura4) positioned in the outer repeats of centromere 1. Inserts that disrupted genes important for heterochromatin activated ura4, and thus the cells were unable to grow when passaged in 5-fluoroorotic acid (FOA) (Figure 2A). Genes with established roles in heterochromatin assembly had significantly fewer insertions in cells with the centromere reporter otr1R::ura4 than in cells lacking the reporter (Figure 2B). The list of candidates consisted of a total of 199 genes and, importantly, 65 are known to be essential for viability. These essential genes were candidates because they tolerated many insertions in their 3′ sequences that reduced heterochromatin but not viability. The high number of essential genes is significant in that most proteins found to be important for heterochromatin are identified in screens of deletion strains that cannot include essential genes. The 199 candidates showed highly significant enrichments for functions in silencing at centromere outer repeats and included all four factors that produce siRNA.
Figure 2. Dense maps of transposable element integration identify genes important for heterochromatin at centromere repeats.
Click image to view.
A. Single insertions of the transposable element Hermes were generated in cells with WT cen1 and cen1 otr1R::ura4. Cultures were passaged in 5-fluoroorotic acid (FOA) for 5 or 80 generations. Cells with insertions in heterochromatin genes (het1) express ura4 and cannot grow in FOA. After growth on FOA fewer insertions were detected in het genes in cells with cen1 otr1R::ura4.
B. Genes involved in forming centromere heterochromatin such as mit1 and sir2 had fewer inserts in cells with the cen1 otr1R::ura4 (black, dupl. libraries) than cells with WT cen1 (red, dupl. libraries).
We identified other RNA–processing factors that were not previously linked to heterochromatin structure. Strikingly, four of the RNA–processing candidates form an interaction module of the canonical mRNA polyadenylation factor and the cleavage factor CPF, as predicted from highly homologous proteins in S. cerevisiae. To determine whether polyadenylation and cleavage contribute to heterochromatin structure at the centromere repeats, we focused on the function of Iss1, a subunit of CPF. We generated a C-terminal truncation of Iss1 (Iss1-ΔC) by removing 38 amino acids that, based on the Hermes insertions, were not important for viability. Iss1-ΔC showed no growth restriction on nonselective medium but exhibited a heterochromatin defect, as demonstrated by growth in the absence of uracil and reduced levels of H3K9 dimethylation (H3K9me2) at otr1R::ura4. The results demonstrated that the Hermes screen correctly identified Iss1 as important for heterochromatin structure at the otr1R::ura4 reporter. Interestingly, we found that Iss1 contributes to the heterochromatin of centromere repeats in cells that lack the otr1R::ura4 reporter but, in this case, the contribution to H3K9me2 was only observed when the RNAi pathway was disabled by deletion of ago1. This role at the outer centromere repeats is therefore independent or redundant with RNAi.
We expanded our study of the Iss1-ΔC mutation to evaluate changes in expression and transcription termination genome-wide. RNA-seq data revealed that Iss1-ΔC did not significantly impact canonical transcription termination, but 73 genes were found to have higher expression. Importantly, the genes overlapped significantly with genes upregulated in cells lacking Rrp6, the 3′-5′ exonuclease subunit of the nuclear exosome. As a key subunit of the nuclear exosome, Rrp6 plays an important role in RNA surveillance in the degradation of meiotic transcripts expressed during vegetative growth and the resulting formation of heterochromatin at these genes. The elimination of meiotic mRNAs depends on the RNA–binding protein Mmi1 to bind to the determinant of selective removal (DSR) sequence in order to recruit the exosome. Our co-immunoprecipitation experiments revealed that Iss1 interacted with Rrp6, Mmi1, and the polyA polymerase Pla1, indicating that Iss1 is associated with this network of elimination factors. Significantly, the interaction with Mmi1 was disrupted by the Iss1-ΔC mutation, a mutation that greatly reduced H3K9me2 at meiotic genes. We tested whether Iss1 plays a direct role in the heterochromatin of meiotic genes by performing ChIP-seq of Iss1-FLAG. While a subset of Iss1–bound genes was highly-expressed and was associated with the canonical function of Iss1 in mRNA termination, most Iss1–bound peaks showed a strong correlation with genes regulated by RNA elimination and heterochromatin. Importantly, the iss1-ΔC mutation caused significant increases in the RNA levels of these genes. Taken together, our studies of RNA levels, Iss1 association with chromatin, and H3K9me2 indicate that Iss1 plays a direct role in the formation of heterochromatin at meiotic genes. Our application of Hermes profiles to identify genes important for heterochromatin formation demonstrates the significance of the approach, especially given that we were able to identify large numbers of essential genes, a result that is not obtainable with other screens.
Retrotransposon insertions associated with risk of neurologic and psychiatric diseases
Neurologic and psychiatric disorders affect 25% of the world population. Given the complexity of the mammalian nervous system, the genetic and cellular etiology of such diseases remains largely unclear. Progress in genetic methodology has provided the potential to identify mechanisms that underlie the diseases. One approach that has successfully identified important disease loci is genome-wide association studies (GWAS). However, in the cases of neurologic and major psychiatric disorders, GWAS have identified large numbers of loci, each associated with small increases in risk. Importantly, there is extensive overlap of the loci that contribute to major psychiatric disorders, indicating that related molecular mechanisms may underlie distinct clinical phenotypes.
The single-nucleotide polymorphisms (SNPs) identified by GWAS to have highest disease association. Trait associated SNPs (TASs) are genetic tags identifying a genomic region that contains the causal mutation(s) leading to the disease risk. Limits on the design of GWAS typically prevent such studies from identifying causal gene alleles. Determining causal variants remains the most challenging and rate-limiting, but also the most important, step in defining the genetic architecture of diseases. The vast majority of GWAS TASs lie in intergenic or intronic regions and therefore do not alter coding sequence. For such SNPs to be causal they would likely have regulatory effects on transcription. Structural variants such as rearrangements, copy number variants, and TE insertions constitute a substantial and disproportionately large fraction of the genetic variants found to alter gene expression.
In humans, the dominant families of TEs are Long INterspersed Element-1 (LINE-1 or L1) and Alu Short Interspersed Elements (SINEs), which are mobilized by L1. TEs alter gene expression particularly easily because they have evolved various sequences that act on enhancers. Given that TEs make up approximately 45% of the human genome, it is not surprising that their regulatory features are abundant sources of tissue-specific promoter activity.
Relatively recent TE insertions can proliferate in the population and become common alleles. The 1000 Genomes Project described genetic variation of diverse human populations by sequencing whole genomes of 2,504 individuals. The extensive survey of genetic variation detected 17,000 polymorphic insertions of TEs, which have the potential to alter gene expression and affect common disease risk. Some TEs have been implicated at disease loci detected by GWAS.
Given the difficulty in identifying genetic variants responsible for neurologic and psychiatric disorders and the regulatory capacity of TEs, we tested whether polymorphic TEs are potential causative variants of such diseases. We analyzed 593 GWAS of neurologic and psychiatric diseases, which in total reported 753 TASs. From the 17,000 polymorphic TEs, we found that 76 were in linkage disequilibrium (LD) with TASs, indicating that the TEs were among the variants with the potential to be causative. We extended our analysis by evaluating each candidate TE for a role in altering expression of proximal genes. In one approach we determined whether polymorphic TEs could disrupt regulatory sequences, as annotated with the epigenomic data of the NIH Roadmap Epigenomics Consortium. Ten of the TE candidates were located in regions of chromatin with active regulatory function in neurologic tissues. We also tested whether the polymorphic TEs were significantly associated with altered expression of proximal genes. By analyzing multi-tissue expression data from GTEx (Genotype-Tissue Expression project), we found that 31 of the TASs linked to TEs were expression quantitative trait loci (eQTLs, loci that seek to identify genetic variants that affect the expression of one or more genes) for adjacent genes showing correlation with altered expression within regions of the brain. These expression data, together with epigenetic and eQTL analyses, indicate that polymorphic TE insertions are important candidates for causing disease risk for Parkinson's, schizophrenia, and amyotrophic lateral sclerosis, on par with other variants at these loci.
Additional Funding
- NICHD Distinguished Scholars Program
Publications
- Esnault C, Lee M, Ham C, Levin HL. Transposable element insertion in fission yeast drives adaptation to environmental stress. Genome Res 2019;29:85-95.
- Lee SY, Hung S, Esnault C, Pathak R, Johnson K, Bankole O, Yamashita A, Zhang H, Levin HL. Dense transposon integration reveals essential cleavage and polyadenylation factors promote heterochromatin formation. Cell Rep 2020;30:2686-2698.
- Grech L, Jeffares DC, Sadée CY, Rodríguez-López M, Bitton DA, Hoti M, Biagosch C, Aravani D, Speekenbrink M, Illingworth CJR, Schiffer PH, Pidoux AL, Tong P, Tallada VA, Allshire R, Levin HL, Bähler J. Fitness landscape of the fission yeast genome. Mol Biol Evol 2019;36:1612-1623.
- van Opijnen T, Levin HL. Transposon insertion sequencing, a global measure of gene function. Annu Rev Genet 2020;54:337-365.
Collaborators
- Jürg Bähler, PhD, University College London, London, United Kingdom
- Shiv Grewel, PhD, Laboratory of Biochemistry and Molecular Biology, NCI, Bethesda, MD
- Stephen Hughes, PhD, Retroviral Replication Laboratory, HIV Drug Resistance Program, NCI, Frederick, MD
- Mamuka Kvaratskhelia, PhD, Ohio State University, Columbus, OH
- Matthew Plumb, BS, Ohio State University, Columbus, OH
- Akira Yamashita, PhD, National Institute for Basic Biology, Okazaki, Japan
Contact
For more information, email henry_levin@nih.gov or visit http://sete.nichd.nih.gov.