Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Lisa Zhang, PhD, Staff Scientist
- Irina Arnaoutova, PhD, Staff Scientist
- Sudeep Gautam, PhD, Visiting Fellow
- Nelson George, PhD, Visiting Fellow
- Javier Anduaga, BS, Pre-Intramural Research Training Award Fellow
- Hung-Dar Chen, PhD, Contract Technician
- Cheol Lee, PhD, Contract Technician
We conduct research to delineate the pathophysiology of and develop novel therapies for the two major subtypes of type I glycogen storage disease (GSD-I): GSD-Ia, and GSD-Ib. GSD-Ia is caused by a deficiency in the liver/kidney/intestine–restricted glucose-6-phosphatase-α (G6Pase-α or G6PC), and GSD-Ib is caused by a deficiency in the ubiquitously expressed glucose-6-phosphate transporter (G6PT or SLC37A4). G6Pase-α is an endoplasmic-reticulum (ER) transmembrane protein that regulates intracellular glucose production by catalyzing the hydrolysis of G6P to glucose and phosphate. The active sites of G6Pase-α face into the ER lumen and depend on G6PT, another ER transmembrane protein, to translocate G6P from the cytoplasm into the lumen. To function, G6Pase-α must couple with G6PT to form a functional G6Pase-α/G6PT complex, which maintains interprandial glucose homeostasis. GSD-Ia and GSD-Ib patients manifest the common metabolic phenotype of impaired glucose homeostasis and the severe long-term complication of hepatocellular adenoma/carcinoma (HCA/HCC). There is no cure for either GSD-Ia or GSD-Ib. We generated animal models of both disorders, which are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy. We also generated several efficacious G6Pase-α– and G6PT–expressing recombinant adeno-associated virus (rAAV) vectors and provided a proof-of-principle gene therapy in murine GSD-Ia and GSD-Ib, which is safe, efficacious, and appropriate for entering clinical trials. In 2018, our rAAV-G6PC/rAAV-co-G6PC vector (US patent #9,644,216; European patent #EP3074510) technology was licensed to Ultragenyx Pharmaceutical Inc. (Novato, CA), who are currently undertaking a phase I/II clinical trial for GSD-Ia (NCT03517085). The initial dose-escalation trial is currently in progress, and no safety issues have been reported to date.
Prevention of hepatic tumor initiation in murine GSD-Ia by gene therapy and activation of tumor-promoting pathways in HCA/HCC
As stated above, the hallmarks of GSD-Ia are impaired glucose homeostasis and a long-term risk of HCA/HCC. Currently, there is no therapy to address HCA/HCC in GSD-Ia. We have shown that rAAV–mediated G6PC gene transfer to young G6pc–/– mice prevents HCA development. It remains unclear whether G6PC gene transfer during tumor development in GSD-Ia can prevent tumor initiation or abrogate pre-existing tumors. We showed that treating liver-specific G6pc–/– (L-G6pc–/–) mice at the tumor-developing stage with rAAV-G6PC restored hepatic G6Pase-α expression, normalized glucose homeostasis, corrected defective hepatic autophagy, and prevented de novo HCA/HCC development. However, gene therapy could not restore G6Pase-α expression in the HCA/HCC lesions and failed to abrogate the pre-existing tumors. We showed that the expression of 11 β-hydroxysteroid dehydrogenase type-1, which mediates local glucocorticoid activation, is down-regulated in HCA/HCC lesions, leading to impairment in the glucocorticoid signaling that is critical for activation of gluconeogenesis. This suggests that down-regulation of local glucocorticoid action in the HCA/HCC lesions may suppress G6Pase-α restoration by gene transfer. Collectively, our data show that rAAV–mediated gene therapy can prevent HCA/HCC initiation in L-G6pc–/–mice at the tumor developing stage, but cannot reduce any pre-existing tumor burden. [Reference 1].
We examined the signaling pathways promoting HCA/HCC development in GSD-Ia. We showed that G6Pase-α deficiency leads to impaired hepatic autophagy, which leads to sustained accumulation of p62, an autophagy-specific substrate that can activate tumor-promoting pathways, including Nrf2, mTORC1, β-catenin, and YAP. Consistently, HCA/HCC lesions in G6Pase-α–deficient livers display increased accumulation of p62 aggregates and phosphorylated p62 along with activation of Nrf2, mTORC1, β-catenin, and YAP signaling. Compared with HCA, HCC lesions displayed higher expression of oncogenes and PKM2, which are critical for tumorigenesis. Our data show that persistent hepatic autophagy impairment activates several tumor-promoting pathways, which contribute to HCA/HCC development in GSD-Ia.
An evolutionary approach to optimizing G6Pase-α activity for gene therapy of GSD-Ia
GSD-Ia is characterized by impaired glucose homeostasis with a hallmark hypoglycemia following a short fast. Previously, we developed rAAV vectors expressing either the wild-type (rAAV-G6PC) or codon-optimized (co) (rAAV-co-G6PC) human G6Pase-α. The rAAV-co-G6PC vector, which contains a 20% change in the native G6PC coding sequence but has a higher potency, is currently being used in a phase I/II clinical trial for human GSD-Ia (NCT 03517085), as mentioned above. While routinely used in clinical therapies, codon-optimized vectors may not always be optimal. Codon-optimization can impact RNA secondary structure, change RNA/DNA protein binding sites, affect protein conformation and function, and alter post-transcriptional modifications, which may reduce potency or efficacy. We therefore sought to develop alternative approaches that could improve the expression yet minimize the impact of sequence changes resulting from broad codon-optimization. The human, dog, mouse, and rat G6Pase-α share 87–91% sequence identity. Intriguingly, in vitro expression assays have routinely shown that the canine G6Pase-α isozyme is significantly more active than human G6Pase-α. We therefore expanded our analysis to compare G6PC genes across the evolutionary tree, seeking potential codon changes that could enhance enzymatic activity of human G6Pase-α. We identified a Ser-298 to Cys-298 substitution naturally found in canine, mouse, rat, and several primate G6Pase-α isozymes, which, when incorporated into human G6Pase-α sequence, markedly enhanced enzymatic activity. Using G6pc–/– mice, we showed that the efficacy of the rAAV-G6PC-S298C vector (US patent #10,415,044; European patent # EP3236984) was three times higher than that of the rAAV-G6PC vector.
We had shown previously that restoring 3% of normal hepatic G6Pase-α activity in G6pc–/– mice prevents HCA/HCC development and that mice harboring less than 3% of normal hepatic G6Pase-α activity are at risk of tumor development. We also showed that G6Pase-α deficiency leads to hepatic autophagy impairment, which can contribute to hepatocarcinogenesis. We therefore undertook a long-term (66-week) preclinical characterization of the rAAV-G6PC-S298C vector in GSD-Ia gene therapy. We showed that the increased efficacy of rAAV-G6PC-S298C allowed G6pc–/– mice treated with a lower dose of this vector to survive long-term. Our study also showed that the rAAV-G6PC-S298C vector, which contains a 2% change in the native G6PC coding sequence, provides equal or greater efficacy to the codon optimization approach, offering a valuable alternative vector for clinical translation in human GSD-Ia [References 2 & 3].
The signaling pathways implicated in impairment of hepatic autophagy in GSD-Ia
G6Pase-α deficiency in GSD-Ia leads to impaired hepatic autophagy, a recycling process important for cellular metabolism and homeostasis. Autophagy can be regulated by several energy-sensing pathways, including SIRT1, FoxO, AMPK, PPAR-α, and mTOR. In 10–day old global G6pc–/– mice, hepatic autophagy impairment was attributed to activation of mTOR and inhibition of AMPK signaling. In other studies, using adult L-G6pc–/– mice, hepatic autophagy impairment was attributed to down-regulation of SIRT1 signaling independent of mTOR. Given that these studies used a different mouse model, the signaling pathways implicated in autophagy deficiency in young G6pc–/– mice remained unclear. We therefore sought to reconcile the reported differences and elucidate the various energy-sensing signaling pathways regulating autophagy in G6pc–/– mice over the first four weeks of life. We showed that impaired SIRT1, FoxO3a, AMPK, and PPAR-α signaling are responsible for autophagy impairment but that mTOR is minimally involved. Hepatic SIRT1 overexpression corrects defective autophagy, restores FoxO3a expression, improves AMPK signaling but fails to normalize impaired PPAR-α expression or metabolic abnormalities associated with GSD-Ia. Importantly, restoration of hepatic G6Pase-α expression in G6pc–/– mice corrects defective autophagy, restores SIRT1/FoxO3a/AMPK/PPAR-α signaling and rectifies metabolic abnormalities. Taken together, the data show that hepatic autophagy impairment in GSD-Ia is mediated by down-regulation of SIRT1/FoxO3a/AMPK/ PPAR-α signaling, with SIRT1 playing a major role [Reference 4].
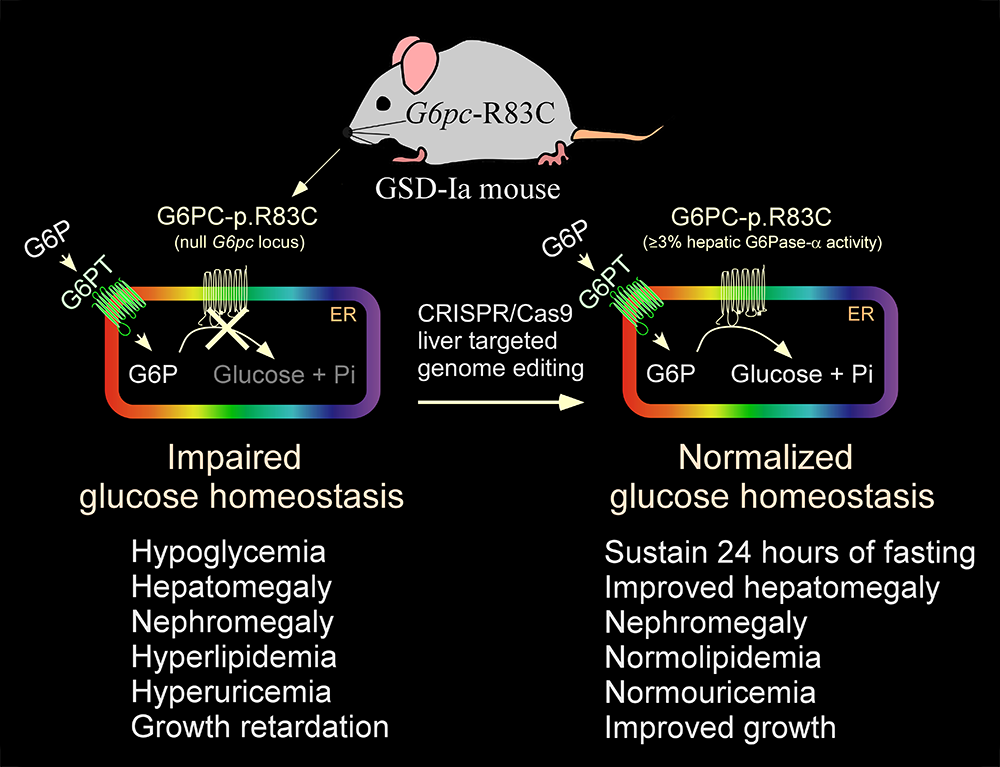
Correction of metabolic abnormalities in a mouse model of glycogen storage disease type Ia by CRISPR/Cas9–based gene editing
The rAAV-G6PC vector used in the current phase I/II clinical trial is episomally expressed, and the long-term durability of expression in humans is currently being established. We therefore sought to explore the use of the CRISPR/Cas9 technology to correct a pathogenic GSD-Ia variant in its native genetic locus. The most prevalent pathogenic mutation identified in Caucasian GSD-Ia patients is G6PC-p.R83C, representing 32% of diseased alleles. Using the CRISPR/Cas9–based gene editing technology, we generated a GSD-Ia mouse disease model, the G6pc-R83C mouse homozygous for the G6PC-p.R83C mutation, and showed that G6pc-R83C mice manifest impaired glucose homeostasis mimicking that of human GSD-Ia. We then used a CRISPR/Cas9–based gene-editing system to treat newborn G6pc-R83C mice and showed that the treated mice grew normally to age 16 weeks without hypoglycemia seizures. The treated G6pc-R83C mice, expressing 3% or more of normal hepatic G6Pase-α activity, maintained glucose homeostasis, displayed normalized blood metabolites, and could sustain 24 hours of fasting. Taken together, we have developed a second-generation therapy in which in vivo correction of a pathogenic G6PC-p.R83C variant in its native genetic locus could lead to potentially permanent, durable, long-term correction of the GSD-Ia disorder [Reference 5].
Figure 1.
Click image to view.
Generation of a mouse model of GSD-Ia and correction of metabolic abnormalities of the GSD-Ia mice by CRISPR/Cas9–based gene editing
Additional Funding
- The Children’s Fund for Glycogen Storage Disease Research
- CRISPR Therapeutics, (Cambridge, MA) under a cooperative research and development agreement (CRADA)
- Beam Therapeutics, Inc (Cambridge, MA) under a CRADA
Publications
- Cho JH, Lee YM, Starost, MF, Mansfield BC, Chou JY. Gene therapy prevents hepatic tumor development in glycogen storage disease type Ia mice at the tumor-developing stage. J Inherit Metab Dis 2019;42:459-469.
- Zhang L, Cho JH, Arnaoutova I, Mansfield BC, Chou JY. An evolutionary approach to optimizing glucose-6-phosphatase-alpha enzymatic activity for gene therapy of glycogen storage disease type Ia. J Inherit Metab Dis 2019;42:470-479.
- Zhang L, Lee C, Arnaoutova I, Anduaga J, Starost MF, Mansfield BC, Chou JY. Gene therapy using a novel G6PC-S298C variant enhances the long-term efficacy for treating glycogen storage disease type Ia. Biochem Biophys Res Commun 2020;527:824-830.
- Gautam S, Zhang L, Arnaoutova I, Mansfield BC, Chou, JY. The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum Mol Genet 2020;29:834-844.
- Arnaoutova I, Zhang L, Chen HD, Mansfield BC, Chou JY. Correction of metabolic abnormalities in a mouse model of glycogen storage disease type Ia by CRISPR/Cas9-based gene editing. Mol Ther 2020;in press.
Collaborators
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genova, Italy
- Youngmok Lee, PhD, University of Connecticut School of Medicine, Farmington, CT
- Brian C. Mansfield, PhD, Foundation Fighting Blindness, Columbia, MD
- Matthew F. Starost, PhD, Diagnostic & Research Services Branch, Division of Veterinary Resources, NIH, Bethesda, MD
- David A. Weinstein, MD, MSc, University of Connecticut School of Medicine, Farmington, CT
Contact
For more information, email chou@helix.nih.gov or visit https://irp.nih.gov/pi/janice-chou.