Protein Sorting in the Endomembrane System

- Juan S. Bonifacino, PhD, Head, Section on Intracellular Protein Trafficking
- Rafael Mattera, PhD, Staff Scientist
- Xiaolin Zhu, Nurse, Technician
- Nireekshit Addanki Tirumala, PhD, Visiting Fellow
- Raffaella De Pace, PhD, Visiting Fellow
- Saikat Ghosh, PhD, Visiting Fellow
- Morié Ishida, PhD, Visiting Fellow
- Ganesh Shelke, PhD, Visiting Fellow
- Adriana Golding, PhD, Intramural Research Training Award Fellow
- Jennifer M. Kunselman, PhD, Intramural Research Training Award Fellow
- Nicole Hsiao-Sánchez, BS, Postbaccalaureate Fellow
- Bob Hsieh, BS, Postbaccalaureate Fellow
- Robert Kaltenbach, BS, Postbaccalaureate Fellow
Our laboratory investigates the molecular mechanisms by which transmembrane proteins (referred to as cargo) are sorted to different compartments of the endomembrane system in eukaryotic cells. The system comprises an array of membrane-enclosed organelles including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs) (e.g., melanosomes, cytotoxic granules), and different domains of the plasma membrane in polarized cells such as epithelial cells and neurons (Figure 1). Transport of cargo between these compartments is mediated by vesicular or tubular carriers that bud from a donor compartment, translocate through the cytoplasm, and fuse with an acceptor compartment. Work in our laboratory focuses on the molecular machineries that mediate these processes, including (1) sorting signals and adaptor proteins that select cargo for packaging into transport carriers (Figure 1), (2) microtubule motors and organelle adaptors that drive movement of transport carriers and other organelles through the cytoplasm, and (3) tethering factors that promote fusion of transport carriers to acceptor compartments. We study these machineries in the context of different intracellular transport pathways, including endocytosis, recycling from endosomes to the plasma membrane, retrograde transport from endosomes to the TGN, biogenesis of lysosomes and LROs, autophagy, and polarized sorting in epithelial cells and neurons. Knowledge gained from this fundamental research is applied to the elucidation of disease mechanisms, including congenital disorders of protein traffic, such as the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS), hereditary spastic paraplegias (HSPs), and other neurodevelopmental disorders.
Figure 1.
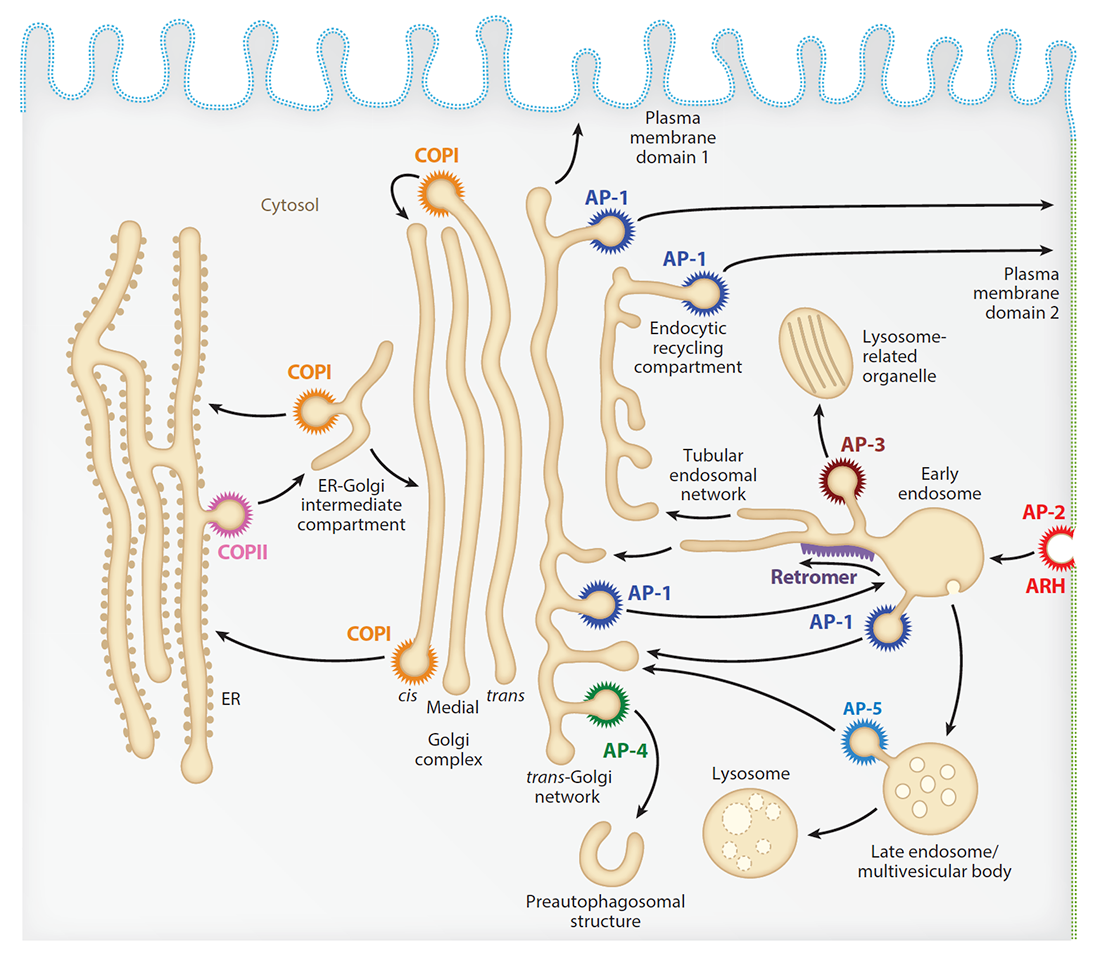
Click image to view.
Schematic representation of the endomembrane system of eukaryotic cells showing the localization of coats involved in protein sorting.
The adaptor protein chaperone AAGAB promotes assembly of the AP-4 complex.
Adaptor protein 4 (AP-4) is a heterotetrameric complex, composed of epsilon, beta4, mu4, and sigma4 subunits, that mediates export of transmembrane cargos, including autophagy protein 9A (ATG9A), from the TGN towards pre-autophagosomal structures (Figure 1). AP-4 has received particular attention in recent years because mutations in any of its subunits cause a complicated form of hereditary spastic paraplegia referred to as “AP-4–deficiency syndrome.” This year, we reported that the alpha- and gamma-adaptin–binding protein (AAGAB, also known as p34) binds to and stabilizes AP-4 subunits, thus promoting complex assembly. The physiological importance of these interactions is underscored by the observation that AAGAB–knockout cells exhibit reduced levels of AP-4 subunits and accumulation of ATG9A at the TGN, like those in cells with mutations in AP-4–subunit genes. The findings demonstrated that AP-4 assembly is not spontaneous but AAGAB–assisted, further contributing to the understanding of an adaptor protein complex that is critically involved in the development of the central nervous system.
ATG9A transport to the cell periphery by RUSC2–mediated coupling to kinesin-1
ATG9A cycles between the TGN in the perinuclear area and pre-autophagosomal structures in the peripheral area of the cell. We recently found that the AP-4 accessory protein RUSC2 couples ATG9A–containing vesicles to the plus-end-directed microtubule motor kinesin-1 via an interaction between a disordered region of RUSC2 and the kinesin-1 light chain (KLC). The interaction is counteracted by the microtubule-associated WD40–repeat domain 47 protein (WDR47). The findings uncovered a mechanism for the peripheral distribution of ATG9A–containing vesicles that involves the function of RUSC2 as a kinesin-1 adaptor and WDR47 as a negative regulator of this function.
ATG9A enables lipid mobilization from lipid droplets.
ATG9A is a scramblase that flips phospholipids between the two membrane leaflets, thus contributing to the expansion of the autophagosome membrane. We found that depletion of ATG9A not only inhibits autophagy but also increases the size and/or number of lipid droplets in human cell lines and in C. elegans (the latter work in collaboration with Andy Golden). Moreover, ATG9A depletion blocks transfer of fatty acids from lipid droplets to mitochondria and, consequently, utilization of fatty acids in mitochondrial respiration. The findings indicate that ATG9A plays a critical role in lipid mobilization from lipid droplets to autophagosomes and mitochondria, highlighting the importance of ATG9A in both autophagic and non-autophagic processes.
Autophagy-associated immune dysregulation in a patient with mutations in ATG9A
We also contributed to a study by Peter Williamson and Erwin Gelfand that concerns a patient with compound heterozygous mutations in ATG9A. The patient exhibited hyperplastic proliferations of T and B cells in lung and brain, and defects in lymphocyte memory-cell populations after developing an infection with the Epstein-Barr virus (EBV). The defects were corrected after treatment with the mTORC inhibitor rapamycin. The results point to a novel role of ATG9A and autophagy in lymphocyte biology and provide an example of how genetic studies may suggest effective specific therapeutic interventions.
SNX19 restricts endolysosome motility through contacts with the endoplasmic reticulum.
In addition to coupling to microtubule motors, interactions with other organelles also regulate the movement of endolysosomes within the cytoplasm. In this regard, we found that the sorting nexin protein SNX19 tethers endolysosomes to the endoplasmic reticulum (ER), reducing their motility and contributing to their concentration in the perinuclear area of the cell. Tethering depends on two N-terminal transmembrane domains that anchor SNX19 to the ER, and a PX domain that binds to phosphatidylinositol 3-phosphate on the endolysosomal membrane. The positioning and movement of endolysosomes within the cell is thus the result of a balance between movement driven by microtubule motors and immobilization by tethering to the ER.
RUFY3 is an ARL8 effector that couples endolysosomes to the microtubule motor dynein-dynactin.
The small GTPase ARL8 associates with endolysosomes, leading to the recruitment of several effectors that couple endolysosomes to kinesins for anterograde transport along microtubules, and to tethering factors for eventual fusion with other organelles. This past year, we identified RUFY3 as a novel ARL8 effector that couples endolysosomes to dynein-dynactin for retrograde transport along microtubules. This function of RUFY3 in retrograde transport contributes to the juxta-nuclear redistribution of endolysosomes upon cytosol alkalinization. The findings highlight the role of ARL8 in the control of not only anterograde but also retrograde endolysosome transport.
Reduced axonal endolysosomal motility in a human iPSC–derived inducible neuronal model of NPC1 disease
Niemann-Pick disease, type C1 (NPC1) is a childhood-onset, lethal, neurodegenerative disorder caused by autosomal recessive mutations in the NPC1 gene, and is characterized by impaired cholesterol homeostasis. Mutations in NPC1 lead to deficient transport and accumulation of unesterified cholesterol in endolysosomal compartments and progressive neurodegeneration. This past year, we contributed to the characterization of a novel human iPSC (induced pluripotent stem cell)–derived, inducible neuronal model of NPC1 developed in the laboratory of Forbes Porter. We found that cholesterol accumulation reduces the motility of axonal endolysosomes in such neurons, and that extraction of cholesterol with 2-hydroxypropyl-beta-cyclodextrin remobilizes them. The findings shed light on the pathological mechanisms contributing to neuronal degeneration in NPC1.
Transcytosis and trans-synaptic retention by postsynaptic ErbB4 underlie axonal accumulation of NRG3.
This past year, we also collaborated with the laboratory of Andrés Buonanno to investigate the mechanisms by which Neuregulin 3 (NRG3) localizes to the axon. We found that pro-NRG3 undergoes proteolytic cleavage by the protease BACE1 at the TGN to generate mature NRG3. Mature NRG3 then emerges on the somatodendritic plasma membrane, from where it is re-endocytosed and anterogradely transported into axons by transcytosis. By a mechanism we call “trans-synaptic retention,” NRG3 then accumulates at presynaptic terminals by stable interaction with its receptor ErbB4 on postsynaptic GABAergic interneurons. We propose that trans-synaptic retention may account for the polarized expression of other neuronal transmembrane ligands and receptors.
PI4P and BLOC-1 remodel endosomal membranes into tubules.
In collaboration with the labs of Cédric Delevoye, Graça Raposo, Daniel Lévy, and colleagues, we found that phosphatidylinositol-4-phosphate (PI4P) and the biogenesis of lysosome-related organelles complex 1 (BLOC-1) govern the formation, stability, and functions of recycling endosomal tubules. BLOC-1 tubulates negatively charged membranes, including those containing PI4P produced by type II PI4-kinases. Reduced PI4-kinase expression impairs the recycling of endosomal cargoes and interferes with the life cycles of intracellular pathogens such as Chlamydia and influenza virus, which depend on endosomal recycling for replication.
Alpha-synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes.
We collaborated with Chiara Zurzolo to demonstrate that alpha-synuclein fibrils affect the morphology of lysosomes and impair their function in neuronal cells. Furthermore, alpha-synuclein fibrils induce peripheral redistribution of lysosomes, increasing their transfer to neighboring cells through tunneling nanotubes. Lysosomal membrane permeabilization (LMP) allows the seeding of soluble alpha-synuclein for production of fibrils in lysosomes of acceptor cells. Lysosomes thus function as a Trojan horse for both seeding and propagation of disease pathology.
Publications
- Keren-Kaplan T, Bonifacino JS. ARL8 relieves SKIP autoinhibition to enable coupling of lysosomes to kinesin-1. Curr Biol 2020 31:540–554.
- Guardia CM, Jain A, Mattera R, Friefeld A, Li Y, Bonifacino JS. RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery. Mol Biol Cell 2021 32:ar25.
- Saric A, Freeman SA, Williamson CD, Jarnik M, Guardia CM, Fernandopulle MS, Gershlick DC, Bonifacino JS. SNX19 restricts endolysosome motility through contacts with the endoplasmic reticulum. Nat Commun 2021 12:4552.
- Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CM, Li Y, Maio M, Golden A, Bonifacino JS. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun 2021 12:6750.
- Mattera R, De Pace R, Bonifacino JS. The adaptor protein chaperone AAGAB stabilizes AP-4 complex subunits. Mol Biol Cell 2022 33:ar109.
Collaborators
- Andrés L. Buonanno, PhD, Section of Molecular Neurobiology, NICHD, Bethesda, MD
- Cédric Delevoye, PhD, Institut Curie, INSERM, Paris, France
- Erwin W. Gelfand, MD, National Jewish Medical and Research Center, Denver, CO
- Andy Golden, PhD, Laboratory of Biochemistry and Genetics, NIDDK, Bethesda, MD
- Daniel Lévy, PhD, Institut Curie, INSERM, Paris, France
- Forbes D. Porter, MD, PhD, Section on Molecular Dysmorphology, Clinical Director, NICHD, Bethesda, MD
- Graça Raposo, PhD, Institut Curie, INSERM, Paris, France
- Peter R. Williamson, MD, PhD, Laboratory of Clinical Immunology & Microbiology, NIAID, Bethesda, MD
- Chiara Zurzolo, MD, PhD, Institut Pasteur, Paris, France
Contact
For more information, email bonifacinoj@helix.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/bonifacino.