Vascular Malformations

- Sarah E. Sheppard, MD, PhD, MST, Head, Unit on Vascular Malformations
- Scott Paulissen, PhD, Research Specialist IV
- Georgia Krikorian, BA, Postbaccalaureate Fellow
- Dhyanam Shukla, BA, Postbaccalaureate Fellow
- Kristina Woodis, BS, Postbaccalaureate Fellow
The primary goal of our translational research group is to develop efficacious therapies for patients with complex lymphatic anomalies. To do this, we seek to understand the molecular etiologies of these complex lymphatic malformations, how these molecular etiologies alter molecular signaling, and how this affects the cellular mechanisms regulating normal lymphatic development. Ultimately, these answers will allow us to develop novel therapies.
Complex lymphatic anomaly is a term that encompasses four different complex lymphatic malformations: central conducting lymphatic anomaly (CCLA), generalized lymphatic anomaly (GLA), Kaposiform lymphangiomatosis (KLA), and Gorham Stout disease (GSD). Patients suffer from symptoms such as pleural effusions, pericardial effusions, ascites, and bone lesions, which can cause significant morbidity and even death. Currently, there is only one medication approved for patients with complex lymphatic anomalies caused by PIK3CA. Similar, precision-medicine approaches are needed for patients with other complex lymphatic anomalies.
Research in our lab will combine patient studies and genomics with the zebrafish model to identify novel therapies. The zebrafish model allows for us to manipulate the genetics rapidly to create patient-based models, image the developing vasculature, understand cellular dynamics in vivo, and perform drug screening.
Figure 1. A bedside-to-bench precision medicine program for lymphatic anomalies
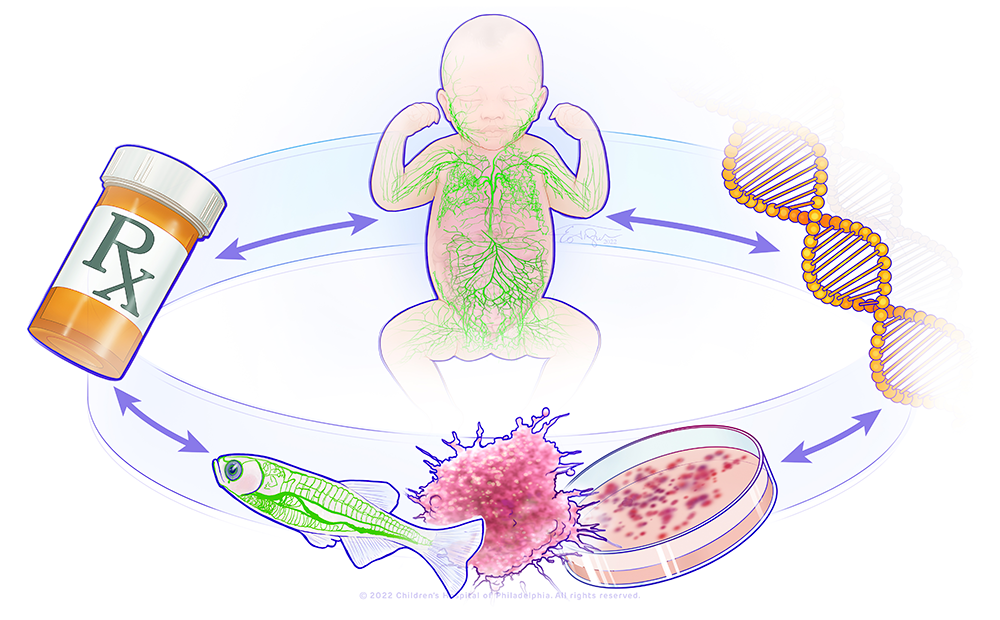
Click image to view.
The figure shows a circle with the major components of the research in the lab: a child with abnormal lymphatics, a DNA molecule, a representation of a zebrafish model and organoid model, and a pill bottle. This represents a bedside-to-bench-to-bedside program using organoid and zebrafish to model patient's lymphatic anomalies and develop therapies that will be translated back to the patients.
Natural history study of lymphatic disorders
We are developing a prospective natural history study for patients with lymphatic anomalies to systematically evaluate the disease phenotypes and long-term outcomes. This will allow us to provide improved prognostication to families, establish screening/monitoring guidelines, determine best practices for genetic diagnosis, and explore family opinions and fertility for those on long-term medication management. The study will allow us to identify novel end-points for future clinical trials.
Genotype-phenotype correlations in central conducting lymphatic anomaly
Central conducting lymphatic anomalies (CCLA) occur when there is a disruption of central lymphatic flow resulting in complications such as non-immune fetal hydrops, chylothorax, chylous ascites, protein-losing enteropathy, other effusions, or lymphedema. The heterogeneity of CCLA complicates diagnosis, treatment, and prognostication. Understanding the molecular etiology of a patient’s disease can drive medical care, including novel treatment strategies. However, few genetic causes have been identified for CCLA. Clinical geneticists use distinct facial features to assist in diagnosis of rare disorders. Given the recent advances in lymphatic imaging, we sought to understand whether we could use features identified by dynamic contrast magnetic resonance imaging for diagnosing CCLA. We discovered that only about a quarter of patients with CCLA have an underlying genetic diagnosis that can be identified by routine clinical evaluation. We also demonstrated that germline RASopathies, mosaic KRASopathies, PIEZO1–related lymphatic dysplasia, and Trisomy 21 have distinct central lymphatic flow phenotypes.
Figure 2. Clinical imaging of lymphatic anomalies according to genotype

Click image to view.
T2 space and dynamic contrast MR lymphangiography (DCMRL) from seven different genotypes, illustrating lymphatic conduction abnormalities.
A. Mosaic BRAF (p.Val600Glu): T2 space shows significant edema in the intercostal, mesentery, and liver lymphatics (left panel) (arrows) that correlates with abnormal perfusion patterns on intrahepatic DCMRL (right). Also note the abnormal lymphatic thoracic vessels with absence of a normal thoracic duct.
B. Mosaic KRAS (p.Gly12Asp): There is edema on T2 space within the mediastinum and lungs (arrows). Patient also with cystic right kidney (asterisk). Intrahepatic DCMRL demonstrates correlation with mediastinal, pulmonary, and supraclavicular edema, with perfusion of dilated lymphatic structures. Of note, this patient has a central thoracic duct (arrow heads), but it was not patent to the venous circulation on ultrasound contrast imaging.
C. Noonan syndrome (PTPN11 p.Gln510His): T2 space imaging demonstrating mediastinal and intercostal edema predominately. With intranodal DCMRL, these areas correlate with abnormal perfusion (arrows). Again, note there is no central thoracic duct, but persistent pulmonary and intercostal perfusion.
D. Trisomy 21: T2 space imaging with edema in the supraclavicular and superior mediastinal lymphatics (arrows). On intrahepatic DCMRL, there is retrograde flow into retroperitoneal lymphatics, intercostal, mediastinal, pulmonary, and supraclavicular perfusion (arrows). There is a patent thoracic duct that courses to the left venous angle (arrowhead).
E. PIEZO1: T2 space shows bilateral pleural effusions and pulmonary and retroperitoneal edema (arrows). Intrahepatic DCMRL shows extensive flow to the hepatic capsular lymphatics, with extension into the mediastinum and pulmonary lymphatics (arrows). There is also retrograde flow into the retroperitoneal lumbar and mesenteric lymphatics. There is a small thoracic duct seen coursing to the left venous angle (arrow head), patent on follow-up imaging.
F. Gaucher’s disease Type III: T2 space notable for ascites. Intrahepatic DCMRL shows retrograde perfusion to retroperitoneal lumbar lymphatics and mesentery (arrows). The thoracic duct is mildly dilated and tortuous as it courses to the left venous angle (arrowhead).
G. Andersen's disease: T2 space imaging with significant ascites, pleural effusions, and anasarca (arrows). With intranodal DCMRL, there is extensive dermal perfusion and dilated retroperitoneal lymphatics. A thoracic duct is present and mildly dilated and tortuous (arrowhead). Figure and caption from [Reference 1].
Cellular and molecular mechanism of lymphatic disorders
Previously, I demonstrated that activating variants in KRAS can drive lymphatic malformations in the zebrafish, which can be treated with MEK inhibitors. We identified several genetic causes of CCLA. We will evaluate these novel potential causes to understand their effect on the cellular and molecular mechanisms driving lymphatic development and identify new therapies.
Additional Funding
- NIH Distinguished Scholars Program
Publications
- Liu M, Smith CL, Biko DM, Li D, Pinto E, O'Connor N, Skraban C, Zackai EH, Hakonarson H, Dori Y, Sheppard SE. Genetics etiologies and genotype phenotype correlations in a cohort of individuals with central conducting lymphatic anomaly. Eur J Hum Genet 2022 30(9):1022–1028.
- Byrne AB, Brouillard P, Sutton DL, Kazenwadel J, Montazaribarforoushi S, Secker GA, Oszmiana A, Babic M, Betterman KL, Brautigan PJ, White M, Piltz SG, Thomas PQ, Hahn CN, Rath M, Felbor U, Korenke GC, Smith CL, Wood KH, Sheppard SE, Adams DM, Kariminejad A, Helaers R, Boon LM, Revencu N, Moore L, Barnett C, Haan E, Arts P, Vikkula M, Scott HS, Harvey NL. Pathogenic variants in MDFIC cause recessive central conducting lymphatic anomaly with lymphedema. Sci Transl Med 2022 4(634):eabm4869.
- Szigety KM, Crowley TB, Gaiser KB, Chen EY, Priestley JRC, Williams LS, Rangu SA, Wright CM, Adusumalli P, Ahrens-Nicklas RC, Calderon B, Cuddapah SR, Edmondson A, Ficicioglu C, Ganetzky R, Kalish JM, Krantz ID, McDonald-McGinn DM, Medne L, Muraresku C, Pyle LC, Zackai EH, Campbell IM, Sheppard SE. Clinical effectiveness of telemedicine-based pediatric genetics care. Pediatrics 2022 150(1):e2021054520.
Contact
For more information, email sarah.sheppard@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/sheppard.