Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Sakshi Chauhan, PhD, Visiting Fellow
- Fuki Kudoh, PhD, Visiting Fellow
- Eunju Lee, PhD, Visiting Fellow
- Keita Saeki, MD, PhD, Visiting Fellow
- Vishal Nehru, PhD, Visiting Fellow
The laboratory is interested in chromatin and gene regulation in innate immunity. We study the role of three nuclear factors, histone H3.3, BRD4, and IRF8. Histone H3.3 is a variant histone that is incorporated into nucleosomes along with transcriptional elongation, an unusual but defining feature of the variant. Most other histones are deposited into nucleosomes during replication. For this reason, H3.3 is thought to be involved in epigenetic memory created by transcription, although experimental evidence for memory formation/maintenance is scant. BRD4 is a bromodomain protein of the BET family, expressed broadly in many cells, from early embryos to adults. Through the bromodomain, BRD4 binds to acetylated histones, not unacetylated histones. BRD4 is thus called a ‘chromatin reader,’ a type of regulatory factor capable of conveying epigenome information to gene expression. Furthermore, BRD4, binds to the elongation factor complex P-TEFb through the C-terminal domain, and drives transcription of many genes by causing RNA polymerase II to move through the gene body, generating nascent mRNA. Many recent reports point out that BRD4 promotes growth of cancer cells, including various blood cancers, by mediating the formation of super-enhancers involved in cell-cycle progression. As we reported in 1990, IRF8 is a DNA–binding transcription factor that plays an essential role in innate resistance to a wide array of pathogens (IRF8's structure is shown in Figure 1A). IRF8 is expressed mostly in cells of the myeloid lineage, including monocytes/macrophages, dendritic cells, and microglia. IRF8 is strongly induced when stimulated by interferons (IFN). In addition, it is upregulated when myeloid cells encounter pathogen-derived molecules and agents produced by stress. In turn, IRF8 activates many genes important for host resistance. IRF8–induced genes include those involved in autophagy and lysosome-mediated pathogen clearance. IRF8 does so by binding to small DNA motifs present in promoter and enhancer regions of the target genes.
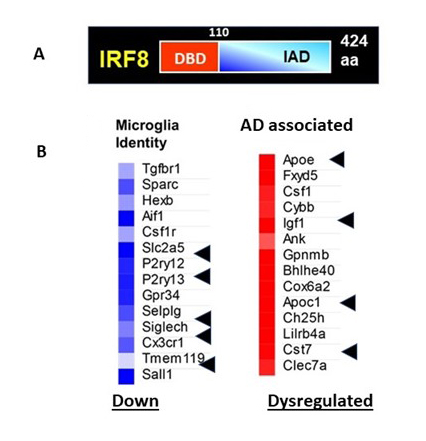
Figure 1. IRF8 regulates microglia transcription
Click image to view.
A. IRF8 domain structure
B. IRF8 directs microglia identity and DAM genes (right). Many genes that define microglia are down regulated in Irf8KO cells (left), while AD associated genes are aberrantly expressed (right).
IRF8 sets microglia-specific epigenome structure and defines the transcriptome program.
Microglia are the only cell type in the brain that protect from pathogen infection and are also important for shaping neuronal development and cellular connections. Recent genome-wide single nucleotide polymorphism (SNP) analyses showed that genetic risk factors for Alzheimer’s disease (AD) are either exclusively or most highly expressed in microglia, not in neurons. It is thought that microglia play a central role in AD onset and progression. Microglia originate from embryonic yolk sac as progenitors, which then migrate into the embryonic brain, where they differentiate into functional microglia in the postnatal stage. In the adult, microglia are distributed throughout the brain, including cortex and hippocampus. Previous reports demonstrated that the transcription factor Spi1 (PU.1) and interferon regulatory factor 8 (IRF8) take part in early progenitor differentiation in the embryonic brain. However, IRF8's function in adult microglia has not been fully understood. We examined microglia from adult brain in Irf8 knockout (KO) mice. We found that Irf8KO microglia have an abnormal morphology and do not express a number of microglia-specific surface markers. We then FACS–sorted microglia from adult wild-type (WT) and Irf8KO mice and performed bulk and single cell RNA-Seq. Our results revealed that, without IRF8, many genes that endow microglia with specific properties were missing or downregulated, including cell-surface markers such as P2ry12, Iba1, Cx3cr1, or Ccr5. On the other hand, some of IFN–stimulated genes (ISGs) and disease-associated microglia (DAM) genes expressed in AD microglia were expressed in Irf8 KO microglia (Figure 1B). In addition, we found that IRF8 is required for the expression of two transcription factors critical for adult microglia function, i.e., Sall1 and Batf3. Our results show that IRF8 directs a transcriptional cascade that defines microglia transcriptome program.
It was important to determine the DNA sites in the microglia genome to which IRF8 binds, information that is missing from the literature. This was technically difficult, because the number of harvestable microglia is low. A modified Cut&Run assay provided reproducible IRF8–binding profiles. Our data showed that IRF8 binds mostly over distant enhancer regions, located upstream and downstream of its target genes (Figure 2). IRF8–binding sites were strongly enriched with DNA motifs containing GAAA. In some enhancers, IRF8 binding was closely clustered, and some sites were within the large stretched enhancers enriched with H3K27ac histone marks. Large enhancers are known to support transcription of genes essential for cell type–specific properties. IRF8 containing large enhancers neighbored genes essential for microglia, including Sall1 and Batf3, transcription factors that direct microglia activity. Deletion of Irf8 led to loss of large enhancers associated with microglia identity genes. Furthermore, an ATAC-Seq assay found that IRF8 is important for setting open chromatin necessary for microglia’s large enhancers. Our results clearly show that IRF8 directs the formation of the microglia-specific epigenome landscape. Consistent with this view, DNA methylome profiles revealed extensive changes in CpG methylation in IRF8KO microglia, indicating that IRF8 regulates overall CpG–island methylation patterns.
Figure 2. IRF8 binds to large enhancers in microglia.

Click image to view.
A. IRF8 distribution in microglia and peritoneal macrophage genome
B. IRF8 binds as clusters colocalizing with H3k27ac and H3K4me1, markers for enhancers.
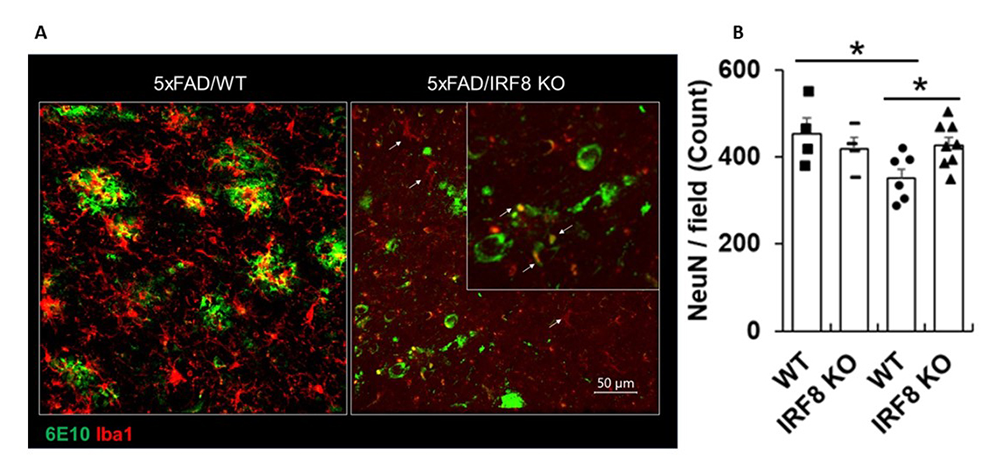
Microglia respond to external immune stimuli such as bacterial lipopolysaccharides (LPS), despite the blood brain barrier, and induce inflammatory genes. We found that IRF8 is required for the expression of many inflammatory genes induced by LPS in microglia. We next sought to determine whether IRF8 impacts the brain’s health. We therefore investigated whether IRF8 affects AD progression, using a mouse model of AD (5xFAD). 5xFAD mice accumulate plaques containing polymerized β-amyloid peptides in the brain near microglia. β-Amyloid plaques are thought to be the main cause of AD pathology, as they lead to neuronal death. We found that 5xFAD mice lacking Irf8 gene have smaller amyloid plaques in the cortex, indicating that IRF8 facilitates plaque formation in this model (Figure 3A). Given that IRF8 shapes microglia identity, our observations are in line with previous reports that microglia worsen AD pathogenesis in the 5xFAD model. NeuN immunostaining found evidence of substantial neuronal loss in the one-year-old 5xFAD brain. However, NeuN immunoreactivity was more intense in Irf8KO 5xFAD brain, indicating reduced neuronal damage in the absence of Irf8 (Figure 3B). These results are consistent with the view that microglia can negatively impact AD pathogenesis
Figure 3. IRF8 modulates Alzheimer's disease progression in mice.
Click image to view.
A. Large amyloid β plaques accumulate near microglia in 5xFAD brain, but amyloid plaques are smaller in Irf8KO 5xFAD brain (cortex). 6E10: polymerized amyloid plaques; Iba1: surface marker for microglia.
B. NeuN immunostaining quantification.
BRD4 promotes cell-cycle progression by preventing DNA damage.
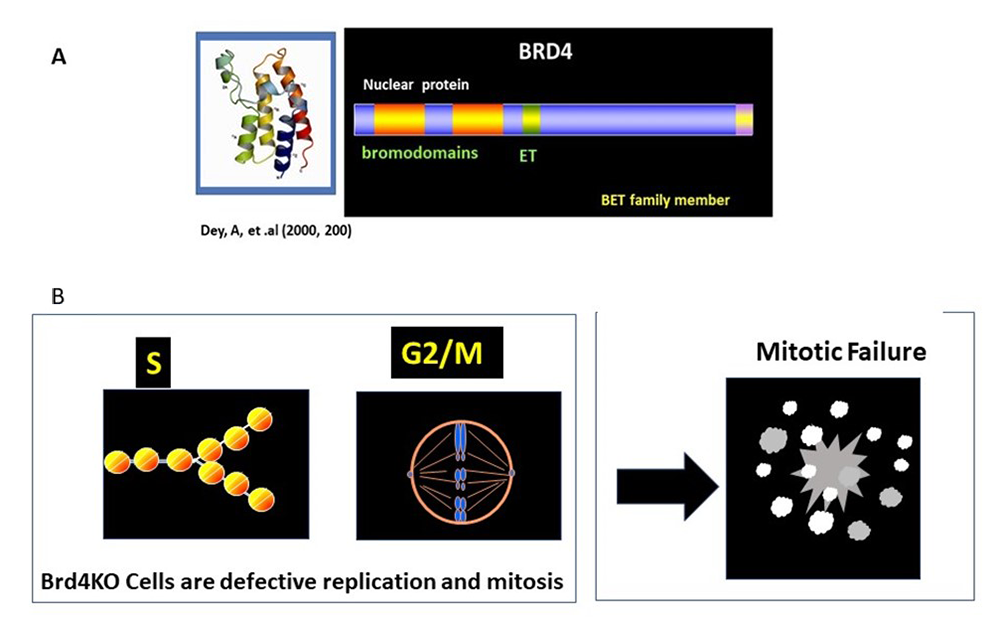
Cell proliferation depends on continuous rounds of cell-cycle progression, which is driven by sequential activation of transcription factors, and other post-translational effectors. Chromatin-binding factor BRD4 is known to promote proliferation of many cancers. BRD4 inhibitors (BETi) can arrest cancer growth (See Figure 4A for BRD4 structure). BETi, and BRD4 Protac inhibitors are thought to represent new therapeutic possibilities against cancer. However, the role of BRD4 in the proliferation of normal cells has remained elusive. We examined proliferation of embryonic fibroblasts from Brd4 conditional knockout (KO) mice. Cell-cycle analysis of WT and Brd4 KO cells showed that BRD4 is required for transition from G0-G1, G1-S, and G2-M (Figure 4B). At G2 to M stage, many Brd4 KO cells underwent catastrophic mitotic failure, as chromosomes failed to align and segregate properly. Transcriptome analysis found that many cell cycle–regulated genes were markedly downregulated in Brd4 KO cells, including several histone genes at S phase, as well as the G2/M master regulators FOXM1 and ATM/ATR. FOXM1 is a transcription factor of the forkhead family and promotes transcription of many G2/M genes. ATM/ATR are kinases previously known to be involved in DNA–damage repair. ATR has been recently shown to be activated at S and necessary for G2-M passage. Our results indicate that BRD4 drives transcription of numerous cell cycle–regulated genes. Consistent with these results, BRD4 occupied numerous cell-cycle genes throughout all stages, as revealed by ChIP-Seq analysis. BRD4 bound to these genes at all stages of cell cycle, seen at the transcription start site (TSS) and gene body.
Remarkably, Brd4KO cells suffered from DNA damage at all stages of cell cycle, which we found by extensive deposition of phospho(γ)–H2AX foci in the nucleus (Figure 4). H2AX is a variant histone H2, phosphorylated upon DNA damage through activation of ATM. Comet assay, another method to detect DNA damage corroborated gH2AX results. DNA damage prompts DNA repair response by a series of enzymes and associated factors, and activates p53. p53 is a central factor, which determines the downstream pathway that causes either cell-cycle arrest or apoptosis. We found that the expression of many DNA–damage control factors, including ATM/ATR, H2AX, and p53, is controlled by BRD4. These results indicate that BRD4 is critical for suppressing endogenous DNA damage, thus playing a major role in the maintenance of genome integrity.
Figure 4. Structure and function of BRD4
Click image to view.
A. BRD4 has two bromodomains and an ET domain.
B. BRD4 controls two fundamental events required for cell growth, replication and mitosis. Brd4KO cells were defective in both and result in mitotic failure.
BRD4 in microglia and neuroinflammation
To further study transcriptome and epigenome regulation of microglia, we began investigating the function of BRD4. We constructed mice in which Brd4 is knocked out in microglia in adult brain (Brd4f/f Cx3cr1Cre), where Brd4 is deleted after Tamoxifen injection. To study how BRD4 regulates neuroinflammation, we analyzed experimental allergic encephalomyelitis (EAE), a mouse model of multiple sclerosis. EAE causes microglia to proliferate from the resident progenitor cells. These microglia, along with T cells and macrophages infiltrating from periphery, promote demyelination and neuronal damage, which results in paralysis. Brain histology revealed that the number of microglia is considerable reduced in Brd4KO brain in EAE (Figure 5A). In addition, Brd4KO microglia exhibited aberrant morphology with stunted extensions and failed to express MHCII, a representative marker for active microglia (Figure 5B). Remarkably, mice with a microglia Brd4 deletion had less demyelination, resulting in reduced tissue damage, and reduced paralysis. Neuro-inflammatory cytokines and chemokines such as interleukin-1β (Il1b) and Ccl were also lower in Brd4KO EAE microglia. These results indicate that BRD4 is required for microglia to detect incoming antigenic and inflammatory signals, interacting with and further stimulating infiltrated T cells. Our study revealed that BRD4 plays a central role in the pathogenesis of neuro-inflammation. It will be of interest to clarify the role of BRD4 in microglia development and AD pathogenesis.
Figure 5. BRD4 limits endogenous DNA damage.

Click image to view.
BRD4 depletion leads to DNA damage. WT cells, Brd4KO cells, and BRD4 inhibitor–treated WT cells were stained with antibody against g-H2AX. Increased DNA damage is verified by quantification on the right.
Figure 6. BRD4 promotes neuroinflammation in the mouse model of multiple sclerosis.

Click image to view.
Brd4KO microglia in EAE. Microglia were visualized by Iba1 immunohistology. The numbers of microglia are reduced in Brd4KO brain (quantification on the right). MHCII expression is absent from Brd4KO microglia. Brd4 deletion in microglia reduces EAE disease phenotypes.
Publications
- Chen L, Ozato K. Innate immune memory in hematopoietic stem/progenitor cells: myeloid-biased differentiation and the role of interferon. Front Immunol 2021 12:621333–621434.
- Murakami K, Sasaki H, Nishiyama A, Kurotaki D, Kawase W, Ban T, Nakabayashi J, Kanzaki S, Ozato K, Tamura T. A RUNX-CBFbeta-driven enhancer directs the Irf8 dose-dependent lineage choice between DCs and monocytes. Nat Immunol 2021 22:301–313.
- Das A, Wang X, Kang J, Coulter A, Shetty AC, Bachu M, Brooks SR, Dell'Orso S, Foster BL, Fan X, Ozato K, Somerman MJ, Thumbigere-Math V. Monocyte subsets with high osteoclastogenic potential and their epigenetic regulation orchestrated by IRF8. J Bone Miner Res 2021 36:199–214.
- Milner JJ, Toma C, Quon S, Omilusik K, Scharping NE, Dey A, Reina-Campos M, Nguyen H, Getzler AJ, Diao H, Yu B, Delpoux A, Yoshida TM, Li D, Qi J, Vincek A, Hedrick SM, Egawa T, Zhou MM, Crotty S, Ozato K, Pipkin ME, Goldrath AW. Bromodomain protein BRD4 directs and sustains CD8 T cell differentiation during infection. J Exp Med 2021 218:e20202512.
- Nakasuji-Togi M, Togi S, Saeki K, Kojima Y, Ozato K. Herbal extracts that induce type I interferons through Toll-like receptor 4 signaling. Food Nutr Res 2022 66:5524. eCollection.
- Kurotaki D, Kikuchi K, Cui K, Kawase W, Saeki K, Fukumoto J, Nishiyama A, Nagamune K, Zhao K, Ozato K, Rocha PP, Tamura T. Chromatin structure undergoes global and local reorganization during murine dendritic cell development and activation. Proc Natl Acad Sci U S A 2022 119(34):e2207009119.
Collaborators
- Steven L. Coon, PhD, Molecular Genomics Core, NICHD, Bethesda, MD
- Robert J. Crouch, PhD, Section on Formation of RNA, NICHD, Bethesda, MD
- Daisuke Kurotaki, PhD, Kumamoto University, Kumamoto City, Japan
- Justin Milner, PhD, University of North Carolina, Chapel Hill, NC
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
- Vivek Thumbigere Math, DDS, PhD, University of Maryland, School of Dentistry, Baltimore, MD
- Jinfang Zhu, PhD, Laboratory of Immunology, NIAID, Bethesda, MD
Contact
For more information, email ozatok@mail.nih.gov or visit https://ozatolab.nichd.nih.gov.