Chromatin Remodeling and Gene Activation

- David J. Clark, PhD, Head, Section on Chromatin and Gene Expression
- Peter Eriksson, PhD, Staff Scientist
- Christopher Coey, PhD, Intramural Research Training Award Fellow
- Hemant K. Prajapati, PhD, Visiting Fellow
- Zhuwei Xu, PhD, Visiting Fellow
- Paul Elizalde, MS, Postbaccalaureate Fellow
- Kayla Gibson, Summer Student (Howard University)
Aberrant gene regulation is the basis of many disease states. Our main objective is to understand how genes are activated for transcription in the context of chromatin structure. Chromatin is not merely a packaging system for DNA in eukaryotic cells; it also participates in gene regulation. The structural subunit of chromatin is the nucleosome, which contains nearly two turns of DNA coiled around a central core histone octamer. The nucleosome is a highly stable structure. Nucleosomes are generally quite regularly spaced along the DNA, like beads on a string (Figure 1). Under physiological of conditions, this nucleosomal filament spontaneously coils to form a heterogeneous chromatin fiber of about 30 nm in diameter (the structure of the fiber is controversial). Chromosomes are composed of functional loops of the chromatin fiber organized by various non-histone proteins. Transcriptionally active and inactive chromatin regions are marked with different sets of post-translational histone modifications. Inactive regions are associated with highly condensed chromatin, as observed in the nucleus and referred to as heterochromatin, whereas active regions are less condensed and referred to as euchromatin. The extreme condensation of heterochromatin is thought to contribute to transcriptional repression. Thus, chromatin restricts access to DNA and is inherently repressive at several structural levels (Figure 1). Given that gene activation requires sequence-specific binding by transcription factors at their cognate sites in gene promoters and at enhancers, the question arises as to how they access their binding sites in chromatin. The answer is provided by the chromatin-remodeling enzymes.
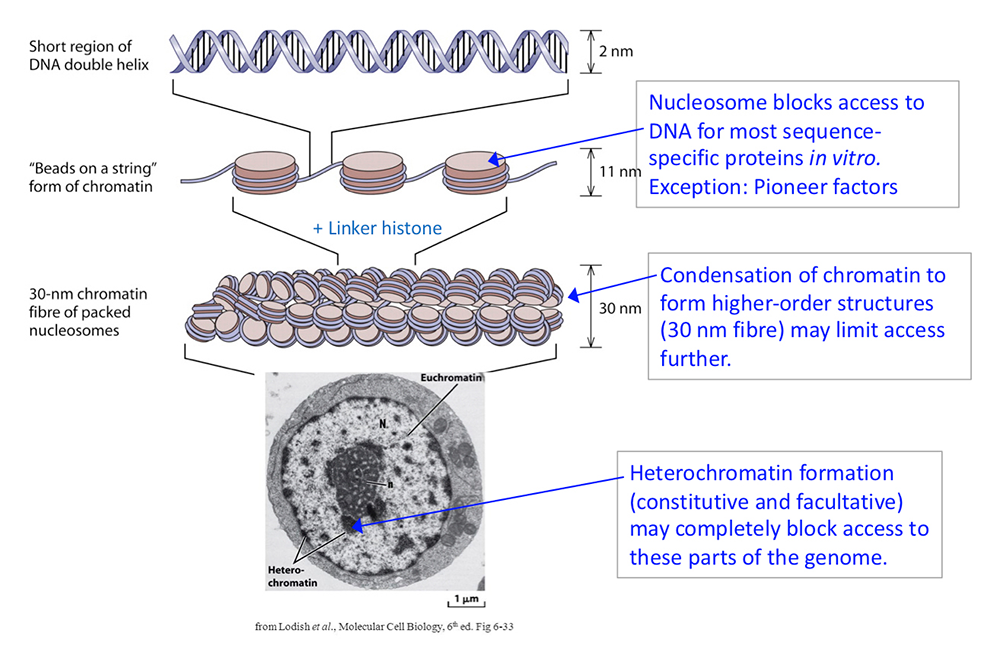
Figure 1. DNA packaging in the nucleus: to what extent does chromatin compaction limit access to the DNA?
Click image to view.
DNA is packaged into the nucleus by histones. The basic structural subunit of chromatin is the nucleosome core, which contains about 147 bp of DNA wrapped nearly twice around a central octamer of core histones. Nucleosomes are regularly spaced along the DNA like beads on a string; the intervening DNA is called the linker DNA and is bound by linker histone (H1). The beads-on-a-string fiber spontaneously condenses into a heterogeneous fiber of about 30 nm width. Genomic regions rich in repetitive elements form constitutive heterochromatin in all cells, in which the chromatin fiber is even more condensed. Facultative heterochromatin is formed on genes that should be permanently silent in a specific differentiated cell type. Heterochromatin is densely packed and darkly staining in the electron micrograph shown here. Euchromatin is less condensed (light staining) and contains active genes. We are interested in determining to what extent chromatin limits DNA accessibility. Figure adapted from Chereji et al. Genome Res 2019;29:1985-1995.
The chromatin-remodeling enzymes can be divided into two groups: histone- or DNA–modifying enzymes, which implement the “epigenetic code”, and ATP–dependent remodeling machines, which move or displace nucleosomes. Histone modifications mark different genomic regions for activation or repression, resulting in recruitment of activating or repressing factors, respectively. Histone-modifying enzymes are typically subunits of large protein complexes containing complementary activities. Similarly, the ATP–dependent chromatin remodelers are usually large complexes containing several subunits, but they use the free energy available from ATP hydrolysis to alter chromatin structure by manipulating nucleosomes. Most of these enzymes can move nucleosomes along DNA, some can form arrays of regularly spaced nucleosomes (e.g., CHD1, ISW1, ISW2, INO80C, SNF2h), while others can remove the histones altogether (e.g., SWI/SNF, RSC, PBAF), or replace histones with variant histones (e.g., SWR1). Human diseases have been linked to defective chromatin-remodeling enzymes. For example, mutations in the hSNF5 subunit of the human SWI/SNF complex are strongly linked to pediatric rhabdoid tumors, and the CHD remodelers have been linked to cancer and autism. Therapies and drugs aimed at epigenetic targets are being tested. Thus, a full understanding of chromatin structure and the mechanisms by which it is manipulated is vital.
Our current objectives are as follows:
- To determine the roles of the major yeast chromatin-remodeling complexes (RSC, SWI/SNF, ISW1, ISW2, CHD1, INO80C) in chromatin organization and gene expression; why there are so many different remodelers and whether they are functionally redundant; so far, our studies indicate that each remodeling enzyme makes a different contribution to chromatin organization;
- to test the hypothesis that nucleosomes control DNA accessibility and play a vital role in gene regulation by blocking promoters.
Sequence-specific transcription factors and DNA accessibility
Gene activation involves the recruitment of a set of factors to a promoter in response to appropriate signals, ultimately resulting in the formation of an initiation complex by RNA polymerase II and transcription, events that coincide with the removal of promoter nucleosomes to create a nucleosome-depleted region (NDR). This observation has led to the generally accepted model that promoter nucleosomes physically block transcription initiation, acting as repressors by preventing access to specific transcription factor binding sites. The nucleosome is a highly stable structure, containing tightly wound DNA that is largely inaccessible to the majority of sequence-specific DNA–binding proteins. Activation occurs if sequence-specific ‘pioneer’ transcription factors are present (these proteins bind to nucleosomal sites with high affinity), and/or if ‘classical’ transcription factors, which are normally blocked by nucleosomes, recruit ATP–dependent chromatin remodelers to move or evict promoter nucleosomes, thus facilitating initiation complex formation.
Although the main effort of the lab focuses on the role of the ATP–dependent chromatin-remodeling enzymes in gene regulation, with particular emphasis on how they may control the accessibility of genomic DNA, we are also interested in the central role of sequence-specific transcription factors. Transcription factors usually recognize consensus DNA–binding sites, mostly containing 4 to 12 base pairs, and in which some bases may be degenerate (e.g., ‘Y’ for pyrimidine [C or T] or ‘R’ for purine [A or G]). The probability of finding such a site in the genome is often much higher than the number of actual binding sites detected empirically, using methods such as ChIP-Seq. Such consensus sites occur not only in regulatory elements, but also inside genes and elsewhere, where they may or may not be functional. The observation that consensus sites often predict far more transcription factor–binding sites than are actually bound in vivo has led to the proposal that consensus sites in non-regulatory regions are not bound because they are blocked by chromatin. However, our recent measurements of DNA accessibility in yeast and mouse nuclei imply that all consensus sites are likely to be accessible in some cells within a population. Such general but limited accessibility predicts detectable binding at all consensus sites, albeit lower than at sites in nucleosome-free DNA. If true, the hypothesis that chromatin prevents binding by transcription factors to consensus sites outside nucleosome-depleted regions is questionable.
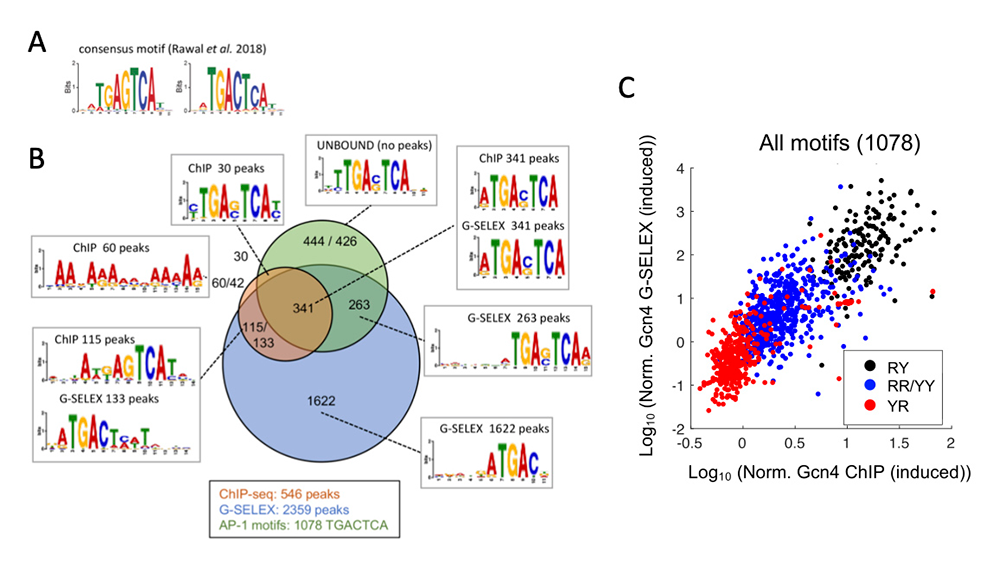
We explored an alternative explanation: that consensus-site sequences derived from ChIP-Seq data may be too degenerate in some cases, such that only a subset of the predicted sites are true sites. We investigated this possibility using the well studied yeast transcription factor Gcn4 as a model [Reference 1]. Previously, we published ChIP-Seq data for Gcn4 in a collaboration with the Hinnebusch lab. In that study, we derived a consensus sequence for Gcn4 binding (Figure 2A), of which there are 1,754 instances in the yeast genome, but only 546 show detectable Gcn4 binding in vivo. To resolve this discrepancy, it is necessary to determine which sites are bound by Gcn4 in the absence of the potential blocking effect of chromatin (i.e., using purified DNA in vitro). Accordingly, we developed a modified SELEX method to identify all sites bound by Gcn4 in the yeast genome, which we termed ‘G-SELEX.’ We used short genomic DNA fragments and purified Gcn4 attached to beads to select DNA fragments containing a Gcn4–bound site. The bound DNA was amplified and incubated with Gcn4 again in a second round of selection; three rounds of selection were performed in total. The final bound product was subjected to paired-end sequencing. We mapped the DNA fragments to the yeast genome to produce a very high-quality coverage map. We identified 2,359 Gcn4–bound sites, but most were bound at very low frequency, and corresponded to Gcn4 half-sites (Figure 2B). In contrast, the major peaks (high-affinity sites) corresponded to the 7–bp sequence TGACTCA. However, of the 1,078 instances of this sequence in the yeast genome, less than half are bound in vitro or in vivo. Further analysis revealed that the bound sites conform to a more extensive consensus: RTGACTCAY, such that RTGACTCAR or YTGACTCAY sites are bound only weakly, and YTGACTCAR sites are not bound in vitro or in vivo (Figure 2C). We conclude that the high-affinity site (RTGACTCAY) essentially accounts for Gcn4 binding in vitro and in vivo, irrespective of whether the site is located in a nucleosome-depleted promoter or inside a gene assembled into nucleosomes.
Figure 2. Sequence-specific binding by the transcription factor Gcn4 can be predicted by its high-affinity consensus site in vitro and in vivo.
Click image to view.
A. Gcn4 consensus binding site derived from ChIP-Seq data.
B. Venn diagram showing the overlap of Gcn4–bound sites in vivo (ChIP-Seq data) and in vitro (G-SELEX data) and the consensus sites derived for each set of sites.
C. Comparison of Gcn4 binding to the 1078 TGACTCA motifs in the yeast genome in vitro and in vivo: high-affinity sites (RY = RTGACTCAY), low-affinity sites (RR = RTGACTCAY and YY = YTGACTGAY), and very poorly bound sites (YTGACTCAR). Note log scale. Figure adapted from [Reference 1].
More generally, we propose that transcription-factor binding sites need to be defined more precisely using quantitative data, which should result in more accurate genome-wide prediction of real binding sites and greater insight into gene regulation. Overall, the study [Reference 1], together with our previous studies, suggests that the prevailing model that chromatin is a general block to gene expression unless specific transcription activators are present may be incorrect. Our current studies are aimed at resolving this issue.
Publications
- Coey CT, Clark DJ. A systematic genome-wide account of binding sites for the model transcription factor Gcn4. Genome Res 2022 32:367–377.
Collaborators
- Jeffrey J. Hayes, PhD, University of Rochester Medical Center, Rochester, NY
- Alan Hinnebusch, PhD, Section on Nutrient Control of Gene Expression, NICHD, Bethesda, MD
- Vasily M. Studitsky, PhD, Fox Chase Cancer Center, Philadelphia, PA
Contact
For more information, email clarkda@mail.nih.gov or visit http://clarklab.nichd.nih.gov.