Pathophysiology, Genetics, and Treatment of Congenital Adrenal Hyperplasia

- Deborah P. Merke, MD, MS, Adjunct Investigator, NICHD, and Chief, Section of Congenital Disorders, Clinical Center
- Deepika D. Burkardt, DO, Staff Clinician
- Rida Javaid, MD, Staff Clinician
- Vipula Kolli, PhD, Staff Scientist
- Qizong Lao, PhD, Staff Scientist
- Elizabeth Joyal, MSN, Nurse Practitioner
- Lee Ann Keener, MSN, Nurse Practitioner
- Sarah Kollender, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Annie Schulman, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Charles Sukin, BS, Postbaccalaureate Intramural Research Training Award Fellow
In its most severe classic form, congenital adrenal hyperplasia (CAH) is a life-threatening, rare orphan disease that is part of the neonatal screen performed in all 50 U.S. states. In its mildest nonclassic form, CAH is one of the most common autosomal recessive diseases and may be a common cause of female infertility. Our research program strives to elucidate the pathophysiology and genetics of CAH, thus facilitating the development of new approaches to the diagnosis, evaluation, and treatment of the disease. We are conducting the largest ever Natural History Study of CAH, with over 450 patients enrolled. We were the first to identify adrenaline deficiency as a new hormonal imbalance in CAH and the first to report smaller-than-normal amygdala, the emotion regulator of the brain, in CAH, providing insight into hormonal effects on the brain. We found that approximately 15 percent of patients with CAH owing to 21-hydroxylase deficiency have a contiguous gene-deletion syndrome resulting in connective tissue dysplasia and a hypermobility-type Ehlers-Danlos syndrome, which represents a novel phenotype named CAH-X. Central to our work is the study of new treatments, including a long-term trial testing sex hormone blockade in children, and novel ways of replacing cortisol, aimed at mimicking the normal circadian rhythm of cortisol secretion. The NIH Clinical Center is the ideal venue in which to carry out such studies and is one of the few places in the world that facilitates the conduct of long-term studies of rare diseases.
Genotype-phenotype studies of CAH-X
CAH is most commonly caused by 21-hydroxylase deficiency. The gene encoding 21-hydroxylase, CYP21A2, and a highly homologous pseudogene, CYP21A1P, map to the short arm of chromosome 6 within the human leukocyte antigen histocompatibility complex. The deleterious sequence in the CYP21A1P pseudogene can be transferred to the CYP21A2 functional gene by homologous recombination, and such events produce common mutations that account for approximately 95% of all CYP21A2 disease–causing mutations. Of the common mutations, approximately 30% are large deletions. The TNXB gene, encoding tenascin-X, an extracellular matrix protein that is highly expressed in connective tissue, and a highly homologous pseudogene, TNXA, flank CYP21A2 and CYP21A1P, respectively. Autosomal recessive tenascin-X deficiency was described as a cause of Ehlers-Danlos syndrome in 2001. We hypothesized that deletions of CYP21A2 might commonly extend into the TNXB gene, and we have been studying this phenomenon in our Natural History Study.
The first evaluation of the potential clinical implications of TNXB heterozygosity in CAH patients was performed in our Natural History Study of CAH (www.ClinicalTrials.gov Identifier No. NCT00250159) at the NIH Clinical Center. In 2013, we prospectively studied 193 consecutive unrelated patients with CAH with clinical evaluations for manifestations of Ehlers-Danlos syndrome and genetic evaluations for TNXB mutations. Heterozygosity for a TNXB deletion was present in 7% of CAH patients; such CAH patients were more likely than age-and sex-matched CAH patients with normal TNXB to have joint hypermobility, chronic joint pain, multiple joint dislocations, and a structural cardiac valve abnormality detected by echocardiography. Six of 13 probands had a cardiac abnormality, including the rare quadricuspid aortic valve, a left ventricular diverticulum, and an elongated anterior mitral valve leaflet. As a result of the study, the term CAH-X was coined to describe the subset of CAH patients who display an Ehlers-Danlos syndrome phenotype resulting from the monoallelic presence of a CYP21A2 deletion extending into the TNXB gene.
The study of CAH-X has provided insight into the recombination events that occur in the class III region of the major histocompatibility complex (MHC) locus, a region in the genome that is predisposed to genetic recombination and misalignment during meiosis. The majority of deletions generate chimeric CYP21A1P/CYP21A2 genes. Chimeric recombination between TNXB and TNXA also occurs (Figure 1). The recombination event deletes CYP21A2 and therefore represents a CAH disease–causing allele. We described three unique types of TNXA/TNXB chimera (CH): CAH-X CH-1 renders the gene nonfunctional, resulting in reduced dermal and serum TNX expression; CAH-X CH-2 alters protein structure; and CAH-X CH-3 is predicted to reduce protein folding energy. Our laboratory continues to investigate how TNXB contributes to the phenotype of CAH patients.
Figure 1. Schematic of CYP21A1P/CYP21A2 and TNXA/TNXB chimeric genes
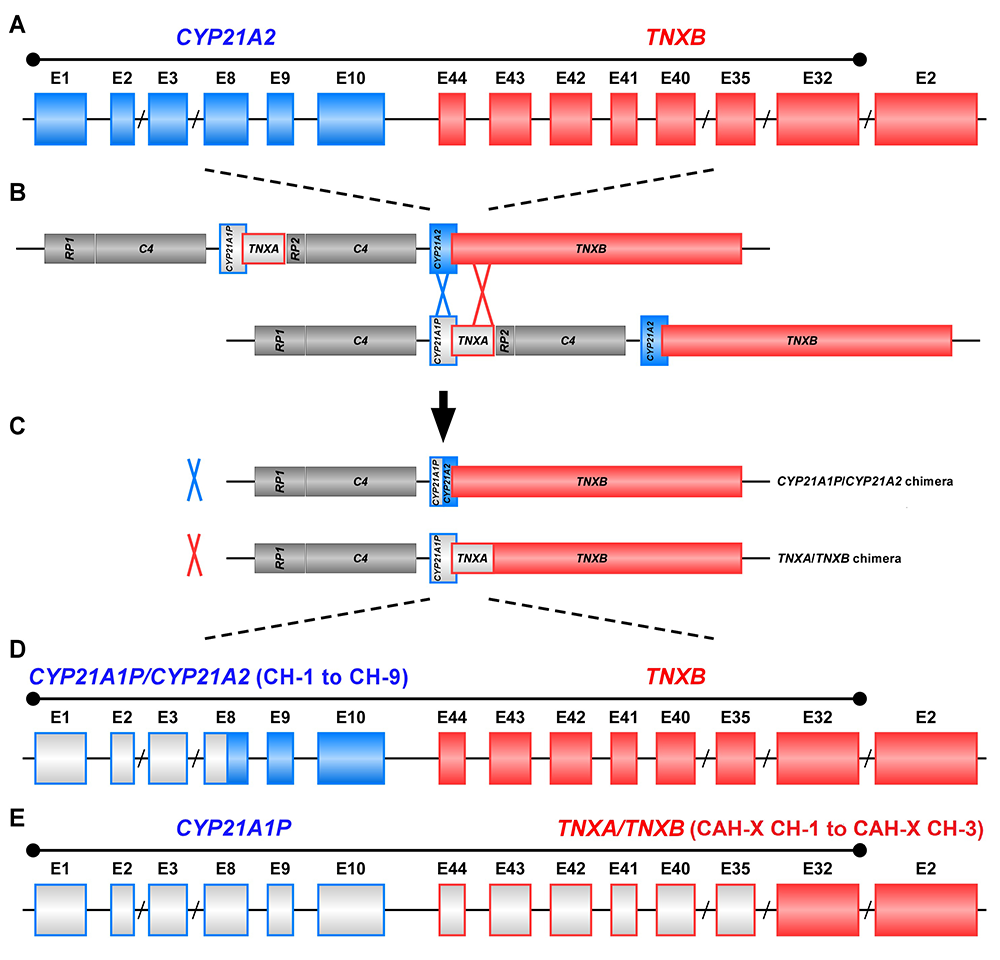
Click image to view.
Formation of chimeric genes occurs as a result of misalignment of homologous genes during meiosis. Active genes are in solid colors; pseudogenes are in grey and are framed with the color of the corresponding functional gene. Representative chimeric genes are shown. In total, there are nine known CYP21A1P/CYP21A2 chimeras (CH-1 to CH-9), and we identified three different types of TNXA/TNXB chimeras (CAH-X CH-1 to CAH-X CH-3) with different junction sites. Approximately 10 percent of patients with CAH owing to 21-hydroxylase deficiency carry at least one TNXA/TNXB chimera, resulting in hypermobility-type Ehlers-Danlos syndrome or CAH-X syndrome.
To date, we have described 24 patients (19 families) with monoallelic CAH-X and three patients with biallelic CAH-X. It is now estimated that approximately 15% of patients with CAH resulting from 21-hydroxylase deficiency are affected by CAH-X. Overall, CAH-X patients have generalized joint hypermobility, subluxations, and chronic arthralgia, and about 25% have cardiac structural abnormalities. Patients with biallelic CAH-X show severe skin hyperextensibility with delayed wound healing and significant joint hypermobility. Other connective-tissue disease manifestations in CAH-X patients include chronic tendonitis and/or bursitis, rectal prolapse, severe gastroesophageal reflux, and cardiac abnormalities. Genetic testing for CAH-X is complex and complicated by pseudogene interference and the large (70kb) size of the TNXB gene. In 2019, we developed a PCR–based, high-throughput, cost-effective assay that accurately identifies CAH-X. The assay had 100% sensitivity and 99.2% specificity.
The study of the CAH-X syndrome provides insight into the complex clinical and genetic characteristics associated with CAH and promises to improve patient outcome through the development of focused medical management aimed at preventing long-term consequences.
Pathophysiology of tumor formation
Patients with CAH are at risk for tumor formation, with the common development of adrenal tumors and adrenal rest tumors (ARTs). ARTs are extra-adrenal-like masses with similarities to adrenocortical tissue. Testicular ARTs (TARTs) are commonly observed in men with CAH, often cause infertility, and are easily detected by ultrasound. The etiology of TARTs and factors contributing to their origin and progression are not completely clear. Some studies support the concept that these benign tumors arise from pluripotent progenitor cells or from cells that are adrenal in origin and which descend with the testis during embryogenesis.
Understanding the pathogenesis and functional features of tumor formation is essential for developing treatment strategies. We performed the first study describing the structural morphology of the cells residing in adrenals from patients with CAH in comparison with ARTs, and we also performed gene-expression studies. We found that CAH–affected adrenal glands and ARTs have similar expression profiles and morphology, demonstrating mostly zona-reticularis characteristics and lymphocytic infiltration, suggesting a common origin that is similarly affected by the abnormal hormonal milieu. The study provided a comprehensive characterization of CAH–affected adrenals and ARTs in relation to control tissues, thus providing insight into disease-specific tissue transformation. In addition, immune system modulators may play a role in tumor formation in CAH.
New and improved biomarkers of CAH
The diagnosis and management of CAH has been limited by inadequate biomarkers. Several pitfalls were identified in the use of 17α-hydroxyprogesterone (17-OHP) and androstenedione, the traditional biomarkers used for disease management. The development of liquid chromatography-tandem mass spectrometry (LC-MS/MS) panels of adrenal steroids has expanded the repertoire of potential new and improved steroid biomarkers. Adrenal-derived 11-oxygenated androgens have emerged as potential new biomarkers, given that traditional biomarkers are subject to variability and are not adrenal-specific, contributing to management challenges. We found that steroids synthesized with the participation of 11beta-hydroxylase (11-oxygenated C19 steroids) are abundant in patients with classic CAH resulting from 21-hydroxylase deficiency, and correlate well with long-term disease control and disease-specific comorbidities (e.g., increased adrenal volume, TARTs, menstrual irregularity, hirsutism) (Figure 2).
Figure 2. Classic and alternative steroidogenesis pathways leading to adrenal androgen production

Click image to view.
In 21-hydroxylase deficiency, elevations of 17α-hydroxyprogesterone and androstenedione can activate alternative steroidogenic pathways (yellow boxes).
With our collaborators Richard Auchus and Adina Turcu, we evaluated 24-hour 11-oxygenated androgens and found diurnal variability in these promising novel biomarkers. We also compared traditional and 11-oxygenated androgens in patients with nonclassic (mild) CAH resulting from 21-hydroxylase deficiency and patients with symptoms of hyperandrogenism from other causes. Patients with nonclassic CAH present with clinical manifestations of hyperandrogenism, features that are shared with other disorders of androgen excess. In particular, the clinical phenotype of women with nonclassic CAH is similar to the more common polycystic ovary syndrome. The diagnosis of nonclassic CAH is based on serum 17-OHP and usually requires dynamic testing with synthetic ACTH (cosyntropin). We found that 11-oxygenated C19 steroids are disproportionately elevated compared with conventional androgens in nonclassic CAH, and that steroid panels can accurately diagnose nonclassic CAH in unstimulated blood tests.
In a retrospective analysis of approximately 2,800 laboratory assessments obtained as part of the Natural History Study of CAH at the NIH Clinical Center, we found discrepant 17-OHP and androstenedione in 469 (17%) of laboratory assessments. Of these, 403 (86%) had elevated 17-OHP with androstenedione in the reference range. Using frozen serum, we evaluated the utility of 11-oxygenated C19 steroids in the setting of inconclusive conventional biomarkers. We found that 11-hydroxytestosterone provided the best discrimination between poor and good clinical control. We continue to explore the utility of these newly described steroids in the diagnosis and management of CAH.
Novel treatment approaches: circadian cortisol replacement
Humans have biological clocks with characteristic patterns of hormone secretion. Cortisol has a circadian rhythm, with levels low at sleep onset, rising between 0200hr and 0400hr, peaking in the early morning, and then declining throughout the day. Existing glucocorticoid replacement is non-physiologic, and the lack of diurnal rhythm may contribute to the many adverse outcomes observed in patients with adrenal insufficiency. In CAH, physiologic cortisol replacement might improve control of adrenal androgens at lower glucocorticoid doses, thus improving patient outcome. A promising treatment approach we have thus been studying is circadian cortisol replacement in patients with CAH.
In 2016, we successfully replaced cortisol in a physiologic manner through the use of a pump usually used to deliver insulin. A programmed 24-hour infusion of hydrocortisone was delivered subcutaneously for six months to eight patients with adrenal insufficiency resulting from 21-hydroxylase deficiency and with multiple comorbidities. Following six months of pump therapy, patients experienced significant improvement in disease control at similar or lower daily doses of glucocorticoid, and significant improvement in their quality of life and fatigue compared with oral conventional therapy. The improvements achieved in androgen control, lean body mass, and health-related quality of life after six months of pump therapy were maintained at eighteen months.
Our group was the first to study circadian cortisol replacement in CAH patients with the use of a modified-release formulation of hydrocortisone, (MR-HC, Chronocort®, CRADA #02800). We successfully completed a phase 2, open-label trial of 16 adults with classic CAH. Compared with various forms of conventional therapy prior to entry, six months of twice daily MR-HC yielded improved disease control throughout the day, using a lower hydrocortisone dose equivalent. Successful completion of this phase 2 study, carried out at the NIH Clinical Center, resulted in a multicenter international phase 3, parallel arm, randomized, open-label study to determine whether this new modified-release preparation of hydrocortisone improves short-term clinical outcome. 122 Adults with classic CAH completed the phase 3 study in 2022. The primary endpoint, 17α-hydroxyprogesterone 24-hour area under the curve standard deviation, did not differ between the two groups; however, improved biochemical control of CAH was observed in the morning and early afternoon in those receiving the MR-HC compared with standard treatments. Sustained benefits with decreased dosage were observed in 18 months extension. Based on these data, MR-HC is now licensed in the UK and Europe. Patients who were enrolled in the phase 3 study are continuing MR-HC therapy in a long-term follow-up study, and future US studies are being planned.
Studies of circadian cortisol replacement provide insight into the role that circadian rhythm plays in the development of the comorbidities associated with adrenal insufficiency. Physiologic cortisol replacement represents a novel treatment approach that promises to improve treatment outcome for patients with CAH, as well as other forms of adrenal insufficiency.
Novel treatment approaches: sex steroid blockade and inhibition
As an alternative approach to the treatment of CAH, the effects of elevated androgen and estrogen could be prevented through the use of sex steroid blockade. Short-term (two-year) administration of an antiandrogen and aromatase inhibitor and reduced hydrocortisone was shown to normalize linear growth rate and bone maturation. A prospective long-term randomized parallel study of the effect of an antiandrogen (flutamide) and an aromatase inhibitor (letrozole), as well as reduced hydrocortisone dose vs. conventional treatment, on adult height is near completion; we will compare data between the treatment groups. The goal of this novel treatment approach is to normalize the growth and development of children with CAH and, ultimately, to determine whether the treatment regimen is effective in improving the growth of children with CAH.
Since the inception of our study of peripheral blockade of sex hormones using an antiandrogen and aromatase inhibitor, new and improved drugs that block sex steroids have been developed. In collaboration with the group of Perrin White, we are studying abiraterone, an irreversible inhibitor of 17α-hydroxylase, a key enzyme required for testosterone synthesis, in a multicenter Phase 1/2 study in prepubescent children (NCT02574910).
Novel treatment approaches: hypothalamic-pituitary-adrenal axis suppression
Potential strategies to address the drivers of excess androgen production in CAH include suppression of the hypothalamic-pituitary-adrenal axis. Newly developed small-molecule antagonists of the corticotropin-releasing factor type 1 (CRF1) receptor antagonist are being studied. In two multicenter, open-label phase II studies of adults with classic CAH, Tildacerfont (Spruce Biosciences, USA) was given at various doses once or twice daily over two weeks and, as a 400 mg dose, for three months. Efficacy analysis was performed based on baseline androstenedione criteria (poor disease control was defined as androstenedione levels more than two times the upper limit of normal and good disease control as less than the upper limit of normal). In comparison with baseline in patients with poorly controlled disease, Tildacerfont treatment induced reductions of disease biomarkers including an average reduction of 74% for circulating levels of ACTH, 82% for levels of 17-hydroxyprogesterone, and 55% for levels of androstenedione. By contrast, ACTH and androgen biomarkers remained stable for patients with good disease control. Overall, Tildacerfont was well tolerated. Multicenter, randomized, double-blind, placebo-controlled, long-term (52 weeks) clinical trials in adults with CAH (NCT04490915, NCT04457336, NCT04544410) are under way to study the effect of CRF-1 antagonists on outcomes such as improved adrenal androgen control, as well as the potential of reducing glucocorticoid dose as an add-on therapy.
Additional Funding
- Cooperative Research and Development Agreement (CRADA) #02800 for Age-Appropriate Hydrocortisone Formulations for the Treatment of Adrenal Insufficiency including Congenital Adrenal Hyperplasia
- NIH U Grant: Abiraterone Acetate in Children with Classic 21-Hydroxylase Deficiency
Publications
- Merke DP, Mallappa A, Arlt W, Brac de la Perriere A, Hirschberg AL, Juul A, Newell-Price J, Perry CG, Prete A, Rees DA, Reisch N, Stikkelbroeck N, Tourraine P, Maltby K, Treasure FP, Porter J, Ross RJ. Modified-release hydrocortisone in congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021 106(5):e2063–e2077.
- Kolli V, Werneck da Cunha I, Kim SA, Iben JR, Mallappa A, Li T, Gaynor A, Coon SI, Quezado MM, Merke DP. Morphologic and molecular characterization of adrenals and adrenal rest affected by congenital adrenal hyperplasia. Front Endocrinol 2021 12:730947.
- Torky A, Sinaii N, Jha S, El-Maouche, Desai J, El-Maouche D, Mallappa A, Merke DP. Cardiovascular disease risk factors and metabolic morbidity in a longitudinal study of congenital adrenal hyperplasia. J Clin Endocrinol Metab 2021 106(12):e5247–5257.
- Mallappa A, Merke DP. Management challenges and therapeutic advances in congenital adrenal hyperplasia. Nat Rev Endocrinol 2022 18(6):337–352.
- Sarafoglou K, Barnes CN, Huang M, Imel EA, Madu IJ, Merke DP, Moriarty D, Nakhle S, Newfield RS, Vogiatzi MG, Auchus RJ. Tildacerfont in adults with classic congenital adrenal hyperplasia: results from two phase 2 studies. J Clin Endocrinol Metab 2021 106(11):e4666–4679.
Collaborators
- Richard J. Auchus, MD, PhD, University of Michigan, Ann Arbor, MI
- Veronica Gomez-Lobo, MD, Pediatric and Adolescent Gynecology, NICHD, Bethesda, MD
- James Marko, MD, Radiology and Imaging Sciences, NIH Clinical Center, Bethesda, MD
- Martha Quezado, MD, Laboratory of Pathology, Center for Cancer Research, NCI, Bethesda, MD
- Richard J. Ross, MD, University of Sheffield, Sheffield, United Kingdom
- Ninet Sinaii, PhD, MPH, Biostatistics and Clinical Epidemiology Service, NIH Clinical Center, Bethesda, MD
- Adina Turcu, MD, University of Michigan, Ann Arbor, MI
- Perrin White, MD, University of Texas Southwestern Medical Center, Dallas, TX
Contact
For more information, email dmerke@nih.gov or visit https://irp.nih.gov/pi/deborah-merke.