Receptors and Actions of Peptide Hormones and Regulatory Proteins in Endocrine Mechanisms

- Maria L. Dufau, MD, PhD, Head, Section on Molecular Endocrinology
- Raghuveer Kavarthapu, PhD, Staff Fellow
- Muruganath Kumar Raju, PhD, Postdoctoral Fellow
- Rajakumar Anbazhagan, PhD, Visiting Fellow
We investigate the molecular basis of peptide hormone control of gonadal function, including the structure and regulation of the genes encoding the luteinizing hormone receptor and prolactin receptor. We also investigate the regulatory mechanism(s) involved in the progression of spermatogenesis and the control of Leydig cell function. Some of this laboratory work focuses on aspects of LHR gene transcription, which is regulated by diverse networks essential for silencing/activation of LHR expression. We also investigate novel gonadotropin-regulated genes relevant to the progression of testicular gametogenesis, Leydig cell function, and other endocrine processes. Major emphasis is placed on studying the function and regulation of the gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), an essential posttranscriptional regulator of spermatogenesis, which was discovered, cloned, and characterized in our laboratory. The various functions of GRTH/DDX25 provide a fertile ground for the development of a non-hormonal male contraceptive.
The luteinizing hormone receptor
The luteinizing hormone receptor (LHR) is expressed primarily in the gonads, where it mediates signals that regulate ovarian and testicular function. The LHR gene is transcriptionally regulated by diverse networks in which coordination and interactions between regulatory effectors are essential for silencing/activation of LHR expression. The proximal Sp1 site of the promoter recruits histone (H) deacetylases and the Sin3A corepressor complex, which contribute to the silencing of LHR transcription. Site-specific acetylation/methylation, induced by the histone deactylase inhibitor trichostatin A (TSA), causes phosphatase release, which serves as a switch for Sp1 phosphorylation, recruitment of the transcription factor II B (TFIIB) and of RNA polymerase II (Pol II), and transcriptional activation. For TSA–mediated LHR transcription, positive coactivator 4 (PC4), recruited by Sp1, acts as its coactivator and has an essential role in the formation/assembly of the preinitiation complex (PIC) and in TFIIB and Pol II recruitment. Further studies demonstrated association between PC4 and acetylated H3 in TSA–induced LHR derepression in MCF7 cells. The recruitment of PC4 to Sp1 and formation of the PC4–Sp1 complex was thus shown to be essential for LHR transcription.
More recently, tandem mass spectrometry (MS/MS) analysis revealed that PC4 associates with the histone variant H3.3, acetylated at several Lys residues. Using an H3.3–specific antibody, immunoprecipitation demonstrated interaction of PC4 with H3.3 induced by TSA, and the presence of the complex PC4–H3.3 at the LHR promoter was demonstrated by reChiP (a sequential chromatin immunoprecipitation protocol). Depletion of endogenous PC4 or H3.3A/B by siRNA (small interfering RNA) caused marked reduction in TSA–induced formation of the complex, its recruitment to the LHR promoter, and transcriptional activation of the LHR gene, which resulted from a reduction in accessibility of the chromatin at the promoter region of the LHR gene. Pull-down studies demonstrated association of H3/H3.3 with GST–PC4 in MCF7 extracts, while we found no direct association with H4. From these and other findings we concluded that PC4 associates with the tetramer via H3 or H3.3. PC4–H3.3 interaction favors acetylation of H3.3, which leads to chromatin accessibility and gene transcription. The findings indicate a critical role of PC4 association with acetylated H3.3 in TSA–induced, Sp1–activated LHR transcription [Reference 1].
Gonadotropin-regulated testicular RNA helicase
Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), discovered in our laboratory, is a testis-specific member of the DEAD-box family of RNA helicases and is essential for the completion of spermatogenesis. It is present in Leydig cells (LC), meiotic (pachytene spermatocytes), and haploid germ cells (round and elongated spermatids) [Reference 4]. Males lacking GRTH are sterile owing to azoospermia, which results from failure of round spermatids to elongate. We demonstrated its participation in the nuclear export/transport of specific mRNAs, the structural integrity of the chromatoid body (CB), and in storage/processing of relevant mRNAs and their transit/association to the actively translating polyribosomes, where GRTH may regulate translational initiation of genes. GRTH is the only family member of the DEAD-box family regulated by hormones. GRTH transcription is stimulated in LCs by LH/cAMP through direct actions of androgen (A)/the A receptor (AR) (autocrine), and, in paracrine fashion, in germ cells through the AR in Sertoli cells. The upstream region of the GRTH gene directs its expression in germ cells and downstream in the LC. Through these regions, A/AR exerts its direct (endogenous) regulation of the GRTH gene in LC, and indirectly in germ cells. Functional binding sites for germ cell nuclear factor (GCNF) present in round spermatids (RS) and spermatocytes (SP) and its regulation by A/AR were identified in the distal region of the GRTH gene, operative selectively in RS. Current knowledge indicates actions of A on GCNF cell-specific regulation of GRTH expression in germ cells (RS). Also, GRTH exerts negative autocrine regulation of GCNF, linking A actions to germ cells through GCNF as an A–regulated transfactor that controls transcription/expression of GRTH.
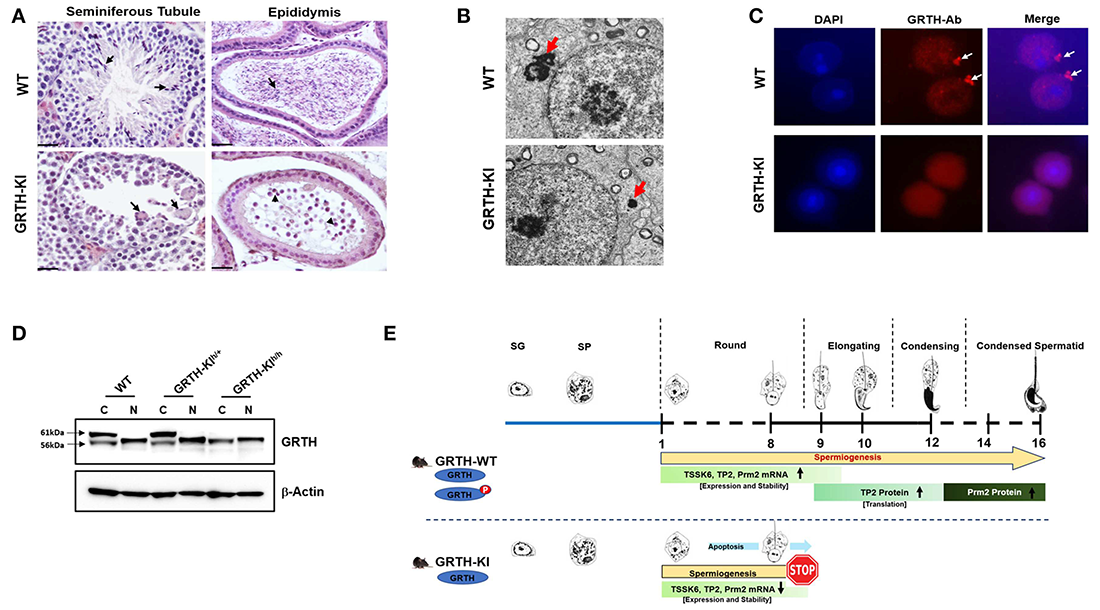
Click image to view.
Figure 1. Essential role of phospho-GRTH (DDX25) in round spermatid during spermiogenesis
A. H&E staining of WT and GRTH–KI mice testis with the human R242H mutation that abolishes phosphorylation of GRTH at T239, showing the seminiferous tubules and epididymis. Lack of pGRTH abolishes the progress of spermatogenesis at step 8 of round spermatids (RS). In WT mice, seminiferous tubules (arrows) contain elongated spermatids, while GRTH–KI mice show the presence of degenerating multinucleated giant cells and the absence of elongated spermatids. The epididymis of WT mice is filled with mature sperm. In contrast, KI mice lack sperm and contain degenerating germ cells.
B. EM sections of testis show marked reduction in the size of the chromatoid body (CB) and of round spermatids (red arrows) in KI compared with WT (CB: non-membranous filamentous cytoplasmic body, which resides in the cytoplasm adjacent to the nucleus of RS and serves as a repository of long-lived mRNAs associated as mRNPs, awaiting translation during spermatogenesis).
C. Immunofluorescence staining of GRTH protein (red) in the nucleus, cytoplasm, and CB of round spermatids of WT mice. GRTH is absent from the CB of KI. Nuclear staining DAPI (blue).
D. Western Blot showing 56 KDa non-pGRTH expression in WT, heterozygous (+/–) and homozygous (–/–) KI mice, while 61 KDa pGRTH was absent from KI–/– mice compared with WT and KI+/– mice.
E. Diagram showing progression of mice spermatogenesis from spermatogonia to RS and the various subsequent steps that give rise to sperm in WT, while in KI–/– mice, owing to the lack of pGRTH, there is a blockade at step 8 of RS. In WT mice, during the process of spermiogenesis, we observed stable expression of Tssk6, TP2, and protamine 2 mRNAs in round spermatids; pGRTH plays a role in the stability of these germ cell–specific mRNAs until they are ready for translation at later steps, during elongation of spermatids. During spermatid elongation, transition proteins are replaced by protamines 1/2, and the chromatin is further condensed. The stability of relevant mRNAs is hampered in KI mice owing to the lack of phospho-GRTH, with a consequent reduction in levels of or abolition of relevant mRNAs expression and degradation [Reference 4].
These findings provide a connection of androgen action to two relevant germ-cell genes (GRTH and GCNF), which are essential for the progress of spermatogenesis, and established their regulatory interrelationship. Our early studies revealed that missense mutation of R to H at amino acid 242 of GRTH, found in 5.8% of patients with complete loss of sperm, causes loss of the 61 kDa phospho-species (pGRTH), with preservation of the 52 kDa nonphospho form. The finding provided an avenue through which to elucidate the function of pGRTH in spermatogenesis. We generated humanized mutant GRTH knock-in (KI) mice. The mice are sterile, with reduced testicular size and lack of sperm (arrest at step 8 of round spermatids [RS]), and demonstrate complete loss of the pGRTH species but with preservation of the nuclear 52 kDa form. The mouse model permits the study of the biological/biochemical functions of the cytoplasmic pGRTH. In KI mice, the nuclear export transport and functions of GRTH are preserved (i.e., mRNA export, miRNA regulation), while the cytoplasmic functions, including shuttling of messages, storage in the CB, and translational events all requiring pGRTH, are absent. We observed marked reduction of the CB size in RS and lack of pGRTH in the CBs. Germ cell apoptosis was present in pachytene spermatocytes (PS) and RS. In contrast to knockout (KO) mice, KI mice showed no changes in miRNA biosynthesis, which excludes participation of pGRTH as transcriptional regulator of the microprocessor complex (the RNase III Drosha and the RNA–binding protein DCGR) that affects primiRNAs formation but indicates the participation of non-phospho GRTH in these processes. In KI mice, there is loss of chromatin remodeling and of related proteins including TP2, PRM2, and TSSK6. Significant decreases of their mRNA and half-lives indicate that their association with pGRTH in the cytoplasm protects these mRNAs from degradation. Also, our work showed that pGRTH stimulates TP2 translation in a 3′ UTR–dependent manner [Reference 2].
In recent studies, we elucidated the GRTH phospho-site at a threonine (Tr239) structurally adjacent to the mutant site found in patients (R242H). Molecular modelling of the phospho-site, based on the RecA domain 1 of the DDX9 crystal structure, elucidated the relevant amino acids that formed the GRTH/PKA interface, solvent accessibility, and H-bonding. In addition to the core residues T239 and R242, these include amino acids E165, K240, and D237 [Reference 3]. Disruption of these relevant amino acids (single or double mutations) caused reduction in or abolition of the p-GRTH at Tr239, which is the cytoplasmic form of GRTH, essential for the progress of spermatogenesis beyond step 8 of round spermatids, with consequent lack of sperm formation. It is worth noting that the deleterious effects on GRTH phosphorylation caused by the mutations did not result from changes of protein kinase A alpha (PKAα) catalytic binding affinity but rather from consequential structural changes that can affect PKA catalytic efficiency. Studies based on the abolition of the phospho-form provide the basis for drug design and virtual and throughput screening for discovery of a reversible chemical inhibitor for use as male contraceptive [Reference 4].
Publications
- Zhao P, Kavarthapu R, Anbazhagan R, Liao M, Dufau ML. Interaction of positive coactivator 4 with histone 3.3 protein is essential for transcriptional activation of the luteinizing hormone receptor gene. Biochim Biophys Acta Gene Regul Mech 2018;1861:971-981.
- Kavarthapu R, Anbazhagan R, Raju M, Morris CT, Pickel J, Dufau ML. Targeted knock-in mice with a human mutation in GRTH/DDX25 reveals the essential role of phosphorylated GRTH in spermatid development during spermatogenesis. Hum Mol Genet 2019;28:2561-2572.
- Raju M, Hassan SA, Kavarthapu R, Anbazhagan R, Dufau ML. Characterization of the phosphorylation site of GRTH/DDX25 and protein kinase A binding interface provides structural basis for the design of a non-hormonal male contraceptive. Sci Rep 2019;9:42857-42867.
- Dufau ML, Kavarthapu R. Gonadotropin regulated testicular RNA helicase, two decades of studies on its structure function and regulation from its discovery opens a window for development of a non-hormonal oral male contraceptive. Front Endocrinol (Lausanne) 2019;10:576-586.
Collaborators
- Sergio A. Hassan, PhD, Center for Molecular Modeling, CIT, NIH, Bethesda, MD
- James M. Pickel, PhD, Transgenic Core Facility, NIMH, Bethesda, MD
Contact
For more information, email dufau@helix.nih.gov or visit http://irp.nih.gov/pi/maria-dufau.