Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Marianne Knue, CRNP, Nurse Practitioner
- Sara Talvacchio, BSN, Research Nurse
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
- Thanh-Truc Huynh, MS, Biologist
- Suman Goshal, PhD, Postdoctoral Visiting Fellow
- Abishek Jha, MBBS, Postdoctoral Visiting Fellow
- Ying Pang, MD, PHD, Postdoctoral Visiting Fellow
- Veronika Caisova, MS, Predoctoral Visiting Fellow
- Ondrej Uher, MSc, Predoctoral Visiting Fellow
- Melissa Gonzales, BS, Postbaccalaureate Fellow
- Karren Antonio, MD, Volunteer
- Felisse Gomez, MD, Volunteer
- Marcela Komrskova, DPharm, Volunteer
- Divya Mamilla, MD, Volunteer
- Mayank Patel, MD, Volunteer
- Ashraf Shoaib, MD, Volunteer
- Isabel Tena Garcia, MD, Volunteer
- Margarita Noreen Valdez, MD, Volunteer
- Boqun Zhu, MD, Volunteer
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare and clinically important chromaffin-cell tumors that typically arise, respectively, from the adrenal gland and from extra-adrenal paraganglia. The clinical features and consequences of PHEO/PGL result from the release of catecholamines (norepinephrine and epinephrine). An undetected PHEO/PGL poses a hazard to patients undergoing surgery, childbirth, or general anesthesia because of the potential for excess catecholamine secretion, which can result in significant, often catastrophic outcomes. Diagnosing and localizing a PHEO/PGL can be challenging. Plasma and urinary catecholamines, as well as their metabolites, and radio-iodinated metaiodobenzylguanidine (MIBG) scanning can yield false-positive/negative results in patients harboring the tumor, and computed tomography (CT) and magnetic resonance imaging (MRI) lack sufficient specificity. The molecular mechanisms by which genotypic changes predispose to the development of PHEO/PGL remain unknown, even in patients with identified mutations. Moreover, in patients with hereditary predispositions, PHEOs/PGLs differ in terms of their growth, malignant potential, catecholamine phenotype, responses to standard screening tests, various imaging modalities, and therefore to different therapeutic options. We focus on developmental, molecular, genetic, epigenetic, proteomic, metabolomic, immunologic, and other types of studies to investigate the bases for predisposition to develop PHEOs/PGLs and the expression of different neurochemical phenotypes and malignant potentials, including therapeutic responses.
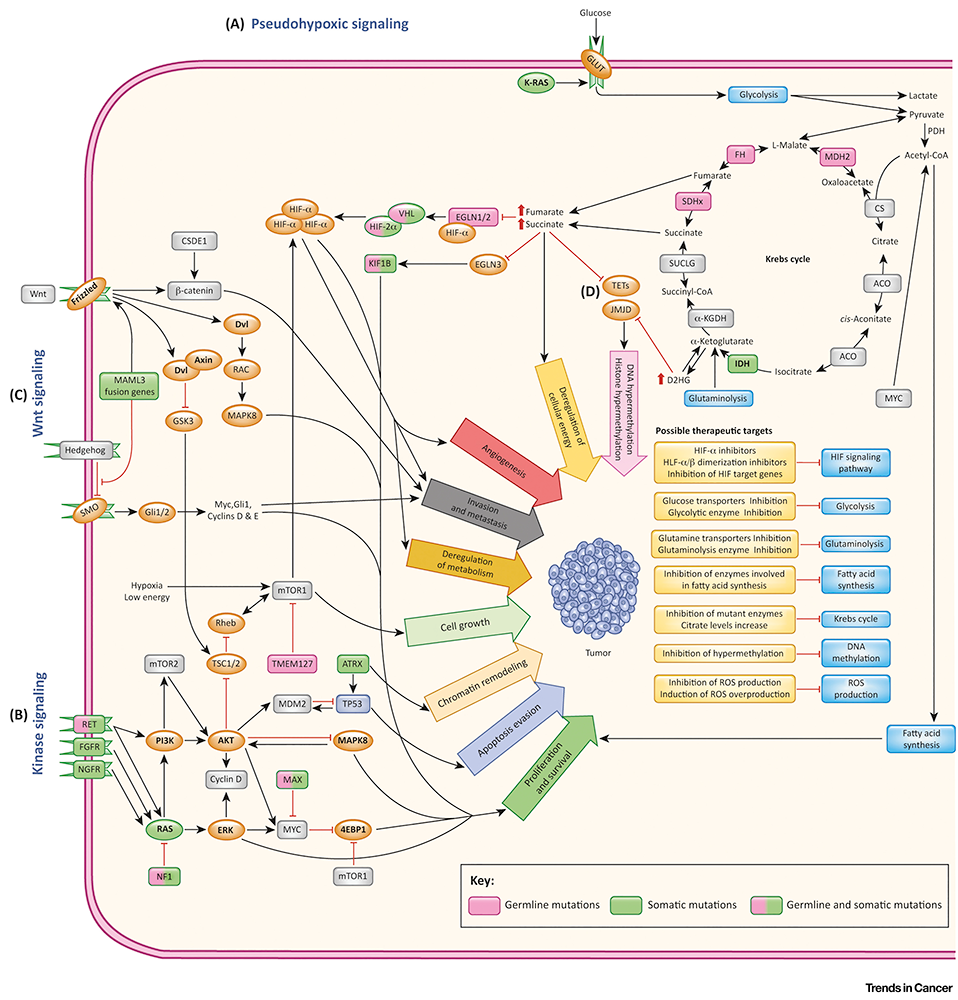
Click image to view.
Figure 1. Genomic landscape of pheochromocytoma and paraganglioma
From: Jochmanova I, Pacak K. Trends Cancer 2018;4(1):6-9.
Clinical and genetic aspects of pheochromocytoma and paraganglioma
Pheochromocytoma and PHEO/PGL can be divided into at least four molecular subgroups. Whether such categorizations are independent factors for prognosis or metastatic disease is unknown. We performed a systematic review and individual patient meta-analysis aimed at estimating whether driver mutation status can predict metastatic disease and survival. Driver mutations were used to categorize patients according to three different molecular systems: two subgroups (SDHB mutated or wild-type [WT]), three subgroups (pseudohypoxia, kinase signaling or Wnt/unknown) and four subgroups (tricarboxylic acid cycle, VHL/EPAS1, kinase signaling or Wnt/unknown). We analyzed 21 studies and 703 patients. Multivariate models for association with metastasis showed correlation with the SDHB mutation, as well as with norepinephrine and dopamine but not with PHEO/PGL location. Other molecular systems were not associated with metastasis. In multivariate models for association with survival, age, and metastases neither paraganglioma nor SDHB mutation remained significant. Other molecular subgroups did not correlate with survival. We concluded that molecular categorization according to SDHB provided independent information on the risk of metastasis. Driver-mutation status did not correlate independently with survival. We believe that these data may ultimately be used to guide current and future risk stratification of PHEO/PGL.
Adrenocortical carcinoma (ACC) and PHEO/PGL are defined by clinico-pathological criteria and can be further subdivided based on various molecular features. Whether differences between these molecular subgroups are significant enough to rechallenge their current clinico-pathological classification is currently unknown. It is also not fully understood to which other cancers ACC and PHEO/PGL show similarity. To address these questions, we included recent RNA-seq data from the TCGA (Cancer Genome Atlas) and TARGET (Therapeutically Applicable Research to Generate Effective Treatments) datasets. Two bioinformatics pipelines were used for unsupervised clustering and principal components analysis and results validated using a consensus clustering model and interpreted according to previous pan-cancer experiments. We studied two datasets consisting of 3319 tumors from 35 disease categories. Consistent with the current classification, ACCs clustered as a homogenous group in a pan-cancer context. It also clustered close to neural crest–derived tumors, including gliomas, neuroblastomas, pancreatic neuroendocrine tumors, and PHEOs/PGLs. By contrast, some PPGLs were mixed with pancreatic neuroendocrine tumors or neuroblastomas. Thus, our unbiased gene-expression analysis of PHEO/PGL did not overlap with their current clinico-pathological classification. The results emphasize the importance of the shared embryological origin of these tumors, all either related or close to neural crest tumors, and open the way for investigation of a complementary categorization based on gene-expression features.
Somatic mutations in hypoxia-inducible factor 2α gene (HIF2A) are associated with the polycythemia-paraganglioma syndrome. Specifically, for the first time, our group described the classic presentation of female patients with recurrent paragangliomas (PGLs), polycythemia (at birth or in early childhood), and duodenal somatostatinomas. Studies demonstrated that somatic HIF2A mutations occur as postzygotic events, while some are associated with somatic mosaicism affecting hematopoietic and other tissue precursors. The phenomenon could explain the development of the early onset of polycythemia in the absence of erythropoietin-secreting tumors. Somatic HIF2A mutations (p.A530V, p.P531S, and p.D539N) were identified in DNA extracted from PGLs of three patients. No somatic mosaicism was detected through deep sequencing of blood genomic DNA. Compared with the classic syndrome, both polycythemia and PGL in all three patients developed at an advanced age, with polycythemia at age 30, 30, and 17 years and PGLs at age 34, 30, and 55 years, respectively. Somatostatinomas were not detected, and two patients had ophthalmic findings. The biochemical phenotype in all three patients was noradrenergic, with 18F-FDOPA PET/CT as the most sensitive imaging modality. All patients demonstrated multiplicity, i.e., lesions in multiple sites, and none developed metastatic disease. The findings suggest that newer techniques need to be developed to detect somatic mosaicism in patients with this syndrome. Absence of HIF2A mosaicism in patients with somatic HIF2A mutations supports association with late onset of the disease, milder clinical phenotype, and an improved prognosis compared with patients who have HIF2A mosaicism.
We also extensively reviewed cardiomyopathies in patients with catecholamine-secreting PHEO/PGL. We also emphasized the necessity of using adrenoceptor blockade in all patients with these tumors. Furthermore, a pharmacologic catecholamine blockade is achieved with α-adrenoceptor and β-adrenoceptor blockers, calcium-channel blockers, or both, with or without the catecholamine synthesis inhibitor metyrosine (Demser). Studies on other medications for catecholamine-induced sinus tachycardia are limited. For the first time, we introduced the use of Ivabradine in the treatment of catecholamine-induced tachyarrhythmia and heart failure. Finally, for the first time we outlined recommended postoperative care of patients with surgically removed PHEO/PGL.
Cancer cells without mitochondrial DNA (mtDNA) do not form tumors unless they reconstitute oxidative phosphorylation (OXPHOS) by mitochondria acquired from host stroma. To understand why functional respiration is crucial for tumorigenesis, we used time-resolved analysis of tumor formation by mtDNA–depleted cells and genetic manipulations of OXPHOS. We showed that pyrimidine biosynthesis dependent on respiration-linked dihydroorotate dehydrogenase (DHODH) is required to overcome cell-cycle arrest, while mitochondrial ATP generation is dispensable for tumorigenesis. Latent DHODH in mtDNA–deficient cells is fully activated with restoration of complex III/IV activity and coenzyme Q redox cycling after mitochondrial transfer, or by introduction of an alternative oxidase. Furthermore, deletion of DHODH interferes with tumor formation in cells with fully functional OXPHOS, while disruption of mitochondrial ATP synthase has little effect. Our results show that DHODH–driven pyrimidine biosynthesis is an essential pathway linking respiration to tumorigenesis, pointing to inhibitors of DHODH as potential anticancer agents.
Imaging of pheochromocytomas and paragangliomas
Phenotyping disease on the basis of nuclear imaging findings heavily depends on genetic background, metabolites, cell membrane specific targets and signaling pathways. PHEOs/PGLs that are related to succinate dehydrogenase subunits mutations (SDHx mutations) are less differentiated than other subgroups and may therefore fail to concentrate 18F-FDOPA, a precursor of catecholamines biosynthesis. However, the 18F-FDOPA–negative phenotype has been reported mostly in SDHx-PHEO/PGL of sympathetic origin, suggesting that both genotype status and location (from sympathetic vs. parasympathetic paraganglia; adrenal vs. extra-adrenal) could influence 18F-FDOPA uptake. The aim of this study was to test whether SDHx drives 18F-FDOPA uptake in the presence of normal epinephrine/norepinephrine concentrations. A cohort of 86 head and neck PHEO/PGL patients (including three metastatic) with normal metanephrines underwent 18F-FDOPA PET/CT. The relationships between 18F-FDOPA uptake and tumor genotype were evaluated. We found that in non-metastatic head and neck PGLs (50 non-SDHx/33 SDHx), no significant difference was observed between these two groups for SUVmax and total lesion uptake. Metastatic head and neck PGLs also had highly elevated uptake values. Our results suggest that neither SDH deficiency nor metastatic behavior influence on 18F-FDOPA uptake in head and neck PGL, probably owing to their high differentiation status, even at metastatic stage. The potential prognosticator value of 18F-FDOPA uptake would need to be further explored in the setting of metastatic PHEO/PGL of sympathetic origin.
An additional multicenter retrospective study included patients from the period 2003 to 2017 with an appropriate CT examination and a histological diagnosis of adrenal adenoma, pheochromocytoma, adrenocortical carcinoma, or metastasis. In total, 346 patients were suitable for the CT image analysis, which included evaluation of the largest diameter, the shape of the lesion, the presence of central necrosis and its margins, and the presence of an enhancing peripheral rim ("ring sign"). PHEOs had a significantly more spherical shape, whereas an elliptical shape significantly reduced the probability of PHEO, as did another shape. A "ring sign" was also more frequent in PHEOs than in other adrenal tumors. A sharp necrosis also increased the probability of PHEO more than unsharp necrosis. The probability calculation model created on the basis of the results confirms a high sensitivity and specificity.
We also summarized recent data related to targeted radionuclide therapy (TRT), which we feel should preferably be performed at specialized centers with an experienced interdisciplinary team. In future, radiotherapy should include the introduction of dosimetry and biomarkers for therapeutic responses for more individualized treatment plans, α-emitting isotopes, as well as the combination of targeted radiotherapies with other systemic therapies.
Immune and metabolic aspects of pheochromocytoma and paraganglioma
Therapeutic options for metastatic PHEO/PGL are limited. We therefore tested an immuno-therapeutic approach based on intratumoral injections of the antibiotic complex mannan-BAM with toll-like receptor ligands into subcutaneous PHEO in a mouse model. The therapy elicited a strong innate immunity-mediated antitumor response and resulted in a significantly lower PHEO volume compared with the phosphate buffered saline (PBS)–treated group and in a significant improvement in mouse survival. The cytotoxic effect of neutrophils, as innate immune cells predominantly infiltrating treated tumors, was verified in vitro. Moreover, the combination of mannan-BAM and toll-like receptor ligands with agonistic anti–CD40 was associated with increased mouse survival. Subsequent tumor rechallenge also supported adaptive immunity activation, reflected primarily by long-term tumor-specific memory. We verified these results further in metastatic PHEO, where the intratumoral injections of mannan-BAM, toll-like receptor ligands, and anti-CD40 into subcutaneous tumors resulted in significantly less intense bioluminescence signals of liver metastatic lesions induced by tail vein injection compared with the PBS–treated group. Subsequent experiments focusing on the depletion of T cell subpopulations confirmed the crucial role of CD8+ T cells in the inhibition of bioluminescence signal intensity of liver metastatic lesions. The results call for a new therapeutic approach in patients with metastatic PHEO/PGL by using immunotherapy that initially activates innate immunity, followed by an adaptive immune response.
Therapeutic aspects of pheochromocytoma and paraganglioma
Neuroendocrine tumors (NETs) express somatostatin receptors, which can be targeted with radiolabeled peptides. In a variety of solid tumors, radio-guided surgery (RGS) has been used to guide surgical resection. 68Ga-DOTA analogs have been shown to be more accurate than other radioisotopes for detecting NETs. A pilot study previously demonstrated the feasibility and safety of 68Ga-DOTATATE RGS for patients with NETs. We evaluated which intra-operative techniques and thresholds define positive lesions that warrant resection during 68Ga-DOTATATE RGS. This prospective cohort study, conducted between October, 2013, and February, 2018, included 44 patients with NETs who underwent 68Ga-DOTATATE RGS. Forty-four patients (22 women and 22 men) had 133 lesions detected on preoperative imaging scans, with a diagnosis of a pancreatic NET (19 of 44), gastrointestinal NET (22 of 44), and PHEO/PGL (3 of 44). The target-to-background ratio (TBR) was obtained by normalizing to the omentum (106 of 133) or other solid organs (27 of 133). The omentum had a significantly lower mean count than other solid organs for background count activity three hours after injection. The lesions containing NETs had a significantly higher TBR than those that did not contain NETs. On a receiver operating characteristic curve analysis, a TBR of 2.5 had a sensitivity of 90% and a specificity of 25%, and a TBR of 16 had a sensitivity of 54% and a specificity of 81%. We concluded that a TBR of 2.5 or greater is a highly sensitive threshold for indicating a lesion to be consistent with a NET on histologic findings and thus warranting surgical resection.
In another study, we investigated the anti-tumor potential of novel molecular-targeted approaches in murine pheochromocytoma cell lines (MPC/MTT), immortalized mouse chromaffin Sdhb–/– cells, 3D-pheochromocytoma tumor models (MPC/MTT spheroids), and human pheochromocytoma primary cultures. We identified the specific PI3Kα inhibitor BYL719 and the mTORC1 inhibitor everolimus as the most effective combination in all models. Single treatment with clinically relevant doses of BYL719 and everolimus significantly decreased MPC/MTT and Sdhb–/– cell viability. A targeted combination of both inhibitors synergistically reduced MPC and Sdhb–/– cell viability and showed an additive effect on MTT cells. In MPC/MTT spheroids, treatment with clinically relevant doses of BYL719 alone or in combination with everolimus was highly effective, leading to a significant shrinkage or even a complete collapse of the spheroids. We confirmed the synergism of clinically relevant doses of BYL719 plus everolimus in human pheochromocytoma primary cultures of individual patient tumors, with BYL719 attenuating everolimus-induced AKT activation. We thus established a method to assess molecular-targeted therapies in human PHEO cultures and identified a highly effective combination therapy. Our data pave the way to customized combination therapy to target individual patient PHEOs/PGLs.
We also continue to work closely with NCI on treating metastatic PHEO/PGL with 177Lu-DOTATATE (Lutathera). Pacak is lead coinvestigator on Frank Lin’s NCI protocol treating these patients.
We wrote a review as a guide for practicing clinicians summarizing current management of PHEO/PGL according to tumor size, location, age of first diagnosis, presence of metastases, and especially underlying mutations in the era of precision medicine.
Animal model of pheochromocytoma and cell culture studies
Our aim was to develop transgenic mice with a gain-of-function Epas1A529V mutation (corresponding to the human EPAS1A530V mutation; EPAS encodes HIF2α) to recapitulate some clinical findings and to discover some new developmental associations with EPAS mutations. We demonstrated elevated levels of erythropoietin and polycythemia, a reduced urinary metanephrine-to-normetanephrine ratio, and elevated expression of somatostatin in the ampullary region of duodenum. Further, inhibition of HIF2α with its specific inhibitor PT2385 significantly reduced erythropoietin levels in the mutant mice. However, polycythemia persisted after PT2385 treatment, suggesting an alternative erythropoietin-independent mechanism of polycythemia. The findings demonstrated the vital roles of EPAS1 mutations in the EPAS1–related syndrome development and the great potential of the Epas1A529V animal model for further pathogenesis and therapeutics studies.
Publications
- El Lakis M, Gianakou A, Nockel P, Wiseman D, Tirosh A, Quezado MA, Patel D, Nilubol N, Pacak K, Sadowski SM, Kebebew E. Radioguided surgery with gallium 68 dotatate for patients with neuroendocrine tumors. JAMA Surg 2019;154:40-45.
- Crona J, Backman S, Welin S, Taieb D, Hellman P, Stalberg P, Skogseid B, Pacak K. RNA-sequencing analysis of adrenocortical carcinoma, pheochromocytoma and paraganglioma from a pan-cancer perspectives. Cancers (Basel) 2018;10:E518.
- Pang Y, Gupta G, Jha A, Yue X, Wang H, Huynh TT, Li A, Baker E, Chew E, Felders RA, Korpershoek E, Zhuang Z, Yang C, Pacak K. Nonmosaic somatic HIF2A mutations associated with late onset polycythemia-paraganglioma syndrome: newly recognized subclass of polycythemia-paraganglioma syndrome. Cancer 2019;125:1258-1266.
- Malaza G, Brofferio A, Lin F, Pacak K. Ivabradine in catecholamine-induced tachycardia in a patient with paraganglioma. New Engl J Med 2019;380:1284-1286.
- Taieb D, Hicks RJ, Hinde E, Guillet BA, Avram A, Ghedini P, Timmers HJ, Scott AT, Elojeimy S, Rubello D, Virgolini IJ, Fanti S, Balogova S, Pandit-Taskar N, Pacak K. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of pheochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019;46:2112-2137.
Collaborators
- Zahraa Abdul Sater, MD, MPH, Clinical Endocrine Section, NIDDK, Bethesda, MD
- James Bibb, PhD, University of Alabama Comprehensive Cancer Center, University of Alabama at Birmingham Medical Center, Birmingham, AL
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Marilo Chiara, PhD, Hospital Universitario Central de Asturias, Oviedo, Spain
- Hans Clevers, PhD, Hubrecht Institute, Royal Netherlands Academy of Sciences, Utrecht, The Netherlands
- Peter Deen, PhD, Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands
- Jaydira Del Rivero, MD, Pediatric Oncology Branch, NCI, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Shane Ellis, PhD, Maastricht Multi-Modal Molecular Imaging Institute, Universiteit Maastricht, Maastricht, The Netherlands
- Stephanie Fliedner, PhD, Universitätsklinikum Schleswig-Holstein, Lübeck Medizinische Klinik I, Lübeck, Germany
- Zdenek Fryšák, MD, PhD, University Hospital and Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czech Republic
- Hans Ghayee, DO, Department of Internal Medicine, University of Florida, Gainesville, FL
- Garima Gupta, MD, Department of Medicine, Jewish Hospital of Cincinnati, Cincinnati, OH
- Peter Herscovitch, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Frank I. Lin, MD, Molecular Imaging Program, NCI, Bethesda, MD
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Renato Mariani-Constantini, MD, PhD, Ageing Research Center (CeSI), D’Annunzio University, Chieti-Pescara, Italy
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Jirí Neužil, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Naris Nilubol, MD, FACS, Surgical Oncology Program, NCI, Bethesda, MD
- Rebecca Oakey, DPhil, King’s College, London, United Kingdom
- Ondrej Petrak, MD, PhD, Third Department of Medicine, General University Hospital, Prague, Czech Republic
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Mercedes Robledo, PhD, Human Cancer Genetics Programme, Spanish National Cancer Centre (CNIO), Madrid, Spain
- Douglas Rosing, MD, Translational Medicine Branch, NHLBI, NIH, Bethesda, MD
- Kelly Roszko, MD, PhD, Skeletal Disorders and Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Hank Stunnenberg, PhD, Faculty of Science, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- David Torpy, MD, PhD, Royal Adelaide Hospital, Adelaide, Australia
- Richard Tothill, PhD, University of Melbourne Centre for Cancer Research, Melbourne, Australia
- Victor Velculescu, MD, PhD, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD
- Brad Wood, MD, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Chunzhang Yang, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Deena Zeltser, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit http://pheopara.nichd.nih.gov.