Transcriptional and Translational Regulatory Mechanisms in Nutrient Control of Gene Expression

- Alan G. Hinnebusch, PhD, Head, Section on Nutrient Control of Gene Expression
- Hongfang Qiu, PhD, Staff Scientist
- Neelam Sen, PhD, Research Fellow
- Swati Gaikwad, PhD, Postdoctoral Fellow
- Suna Gulay, PhD, Postdoctoral Fellow
- Neha Gupta, PhD, Postdoctoral Fellow
- Priyanka Mittal, PhD, Postdoctoral Fellow
- Poonam Poonia, PhD, Postdoctoral Fellow
- Priyanka Singh, PhD, Postdoctoral Fellow
- Anil Thakur, PhD, Postdoctoral Fellow
- Vishalini Valabhoju, PhD, Postdoctoral Fellow
- Anil Vijjamarri, PhD, Postdoctoral Fellow
- Qiaoyun Zheng, PhD, Postdoctoral Fellow
- Jinsheng Dong, PhD, Senior Research Assistant
- Fan Zhang, MS, Senior Research Assistant
- Laura Marler, BS, Graduate Student
We study the fundamental mechanisms involved in the assembly and function of translation initiation complexes for protein synthesis, using yeast as a model system in order to exploit its powerful combination of genetics and biochemistry. The translation initiation pathway produces an 80S ribosome bound to mRNA, with methionyl initiator tRNA (tRNAi) base-paired to the AUG start codon. The tRNAi is recruited to the small (40S) subunit in a ternary complex (TC) with the GTP–bound eukaryotic initiation factor eIF2 to produce the 43S preinitiation complex (PIC) in a reaction stimulated by eIFs 1, 1A, 3, and 5. The 43S PIC attaches to the 5′ end of mRNA, facilitated by the cap-binding complex eIF4F (comprising eIF4E, eIF4G, and the RNA helicase eIF4A) and poly(A)–binding protein (PABP) bound to the poly(A) tail, and scans the 5′ untranslated region (UTR) for the AUG start codon. Scanning is promoted by eIFs 1 and 1A, which induce an open conformation of the 40S and rapid TC binding in a conformation suitable for scanning successive triplets entering the ribosomal P site (P-out), and by eIF4F and other RNA helicases, such as Ded1, that remove secondary structure in the 5′ UTR. AUG recognition evokes tighter binding of the TC in the P-in state and irreversible GTP hydrolysis by eIF2, dependent on the GTPase–activating protein (GAP) eIF5, releasing eIF2-GDP from the PIC, with tRNAi remaining in the P site. Joining of the 60S subunit produces the 80S initiation complex ready for protein synthesis. Our current aims in this research area are to (1) elucidate functions of eIF1, eIF5, eIF3, and 40S proteins in TC recruitment and start codon recognition; (2) identify distinct functions of RNA helicases eIF4A (and its cofactors eIF4G/eIF4B), Ded1, and Dbp1, and poly(A)–binding protein (PABP) in mRNA activation, 48S PIC assembly, and scanning in vivo; (3) uncover the mechanisms of translational repression by the repressors Scd6, Pat1, and the helicase Dhh1; (4) elucidate the in vivo functions of yeast orthologs of eIF2D and the MCT-1/DENR complex in 40S ribosome recycling at stop codons and reinitiation in 3′ untranslated regions in vivo; and (5) elucidate the roles of yeast orthologs of eIF2A and eIF2D in eIF2–independent initiation of translation in stress conditions.
We also analyze the regulation of amino acid–biosynthetic genes in budding yeast as a means of dissecting fundamental mechanisms of transcriptional control of gene expression. During amino acid limitation, transcription of these genes is coordinately induced by the activator Gcn4 as the result of induction of Gcn4 at the translational level. The eviction of nucleosomes that occlude promoter DNA sequences and block access by RNA polymerase is thought to be a rate-limiting step for transcriptional activation. Previous studies implicated certain histone chaperones, ATP–dependent chromatin-remodeling complexes, or histone acetyltransferase (HAT) complexes in eviction of promoter nucleosomes at certain yeast genes, but it is unclear whether these co-factors function at Gcn4 target genes. Our aim is to elucidate the full set of co-factors that participate in promoter nucleosome eviction at Gcn4 target genes, their involvement in this process genome-wide, and the transcriptional consequences of defective nucleosome eviction. Functional cooperation among the chromatin-remodeling complexes SWI/SNF, RSC, and Ino80, as well as the HAT complexes SAGA, NuA4, NuA3, and Rtt109/Asf1, in these processes are under study. We also recently discovered that Gcn4 can activate transcription from binding sites within the coding sequences (CDS) of its target genes, inducing internal subgenic sense and antisense (AS) transcripts in addition to the conventional full-length transcripts that initiate 5′ of the CDS; and we are probing both the mechanism and possible regulatory functions of these internal AS transcripts [Rawal Y, et al. Genes Dev 2018;32:695].
eIF1 interactions with Met-tRNAi and eIF2β control the accuracy of start-codon selection by the scanning preinitiation complex.
As described above, AUG recognition evokes rearrangement from an open PIC conformation with the TC in a P-out state to a closed conformation, with the TC more tightly bound in the P-in conformation. Factor eIF1 binds to the 40S subunit and exerts a dual role of enhancing TC binding to the open PIC conformation while antagonizing the P-in state, necessitating eIF1 dissociation for start codon selection to proceed. Our previous cryo-EM structures of partial yeast PICs revealed juxtaposition of eIF1 Loop 2 with the Met-tRNAi D loop in the P-in state and predict a distortion of Loop 2 from its conformation in the open complex to avoid a clash with Met-tRNAi. We showed that Ala substitutions in Loop 2 increase initiation at both near-cognate UUG codons and AUG codons in poor context in vivo. Consistently, the D71A-M74A double substitution stabilizes TC binding to 48S PICs reconstituted with mRNA harboring a UUG start codon, without affecting eIF1 affinity for 40S subunits. Similar but relatively stronger reductions in discrimination against poor start codons were conferred by arginine substitutions in Loop 2; and none of the Loop 2 substitutions perturbed the rate of TC loading on scanning 40S subunits in vivo. The findings indicate that electrostatic and steric clashing between the eIF1 Loop 2 and tRNAi D loop impede Met-tRNAi accommodation specifically in the P-in state of the closed complex without influencing the P-out mode of TC binding to the open complex; and Arg substitutions convert the Loop 2–tRNAi clash to an electrostatic attraction that stabilizes P-in and enhances selection of poor start codons in vivo. Thus, in contrast to the eIF1A N-terminal tail (NTT), which specifically stabilizes the closed/P-in state of the PIC and enables recognition of poor start codons, eIF1 Loop 2 destabilizes the P-in state and helps ensure relatively greater initiation frequencies for optimal start codons in vivo [Thakur A, Hinnebusch AG. Proc Natl Acad Sci USA 2018;115:E4159].
The β-subunit of eIF2 interacts with eIF1, eIF1A, and the anticodon stem of tRNAi exclusively in the open complex and thus should exclusively stabilize the scanning conformation of the PIC. Supporting this assumption, eIF2β and eIF1 substitutions designed to weaken their mutual interaction increase UUG initiation in vivo and stabilize TC binding at UUG codons in reconstituted PICs in vitro. Moreover, compound substitutions at the interface additionally derepress GCN4 translation, signifying a reduced rate of TC loading to the PIC, which was also reconstituted in vitro. Such genetic and biochemical phenotypes indicate destabilization of the open complex and a shift to the closed/PIN state. Remarkably, an eIF1 substitution designed to strengthen, not weaken, the eIF2β:eIF1 interface had the opposite genetic and biochemical phenotypes. The position of eIF2β in the open complex is also predicted to clash with Met-tRNAi in the closed/PIN state, and substitutions designed to diminish this clash increased UUG initiation in vivo and stabilized Met-tRNAi binding at UUG codons in vitro, but they did not confer a Gcd– phenotype and had little effect on TC loading in vitro, thus showing that eIF2β’s clash with Met-tRNAi disfavors transition to the closed complex without affecting TC binding to the open complex. In summary, eIF2β resembles eIF1 in both (1) stabilizing the open/POUT conformation by its direct binding to eIF1 and Met-tRNAi, and (2) impeding transition to PIN at non-AUG codons through a clash with tRNAi.
Reconstitution of RNA helicase Ded1 function in eIF4F/eIF4A–dependent acceleration of 48S PIC assembly on structured native mRNAs
RNA helicases eIF4A and Ded1 are believed to resolve mRNA structures that impede ribosome attachment or scanning to the start codon, but whether they perform distinct functions in vivo has not been unequivocally established. Previously, we compared the effects of mutations in Ded1 or eIF4A on genome-wide translational efficiencies (TEs) by ribosome profiling. Despite similar reductions in bulk translation, inactivation of Ded1 substantially reduced the relative TEs of more than 600 mRNAs, whereas inactivation of eIF4A affected less than 40 mRNAs in a similar manner. Ded1–dependent mRNAs show greater than average 5′ UTR lengths and propensity for secondary structures, implicating Ded1 in scanning though structured 5′ UTRs. We measured the kinetics of 48S PIC assembly in the yeast reconstituted system for the native mRNAs that we identified as being Ded1 hyper- or hypodependent in vivo by ribosome profiling. Whereas eIF4A was essential for 48S PIC assembly on all mRNAs tested, Ded1–hypodependent mRNAs could be recruited rapidly without Ded1, and addition of Ded1 only moderately accelerated their recruitment. Ded1–hyperdependent mRNAs, by contrast, were recruited poorly in the absence of Ded1, and Ded1 greatly accelerated their recruitment. Eliminating stem-loop (SL) structures in the 5′ UTRs enhanced Ded1–independent recruitment, and diminished Ded1 acceleration of 48S assembly, on several hyperdependent mRNAs. Inserting SLs into a synthetic unstructured 5′ UTR conferred a strong Ded1 requirement for rapid recruitment. Previous biochemical findings indicated that Ded1 unwinding activity is stimulated by its interactions with eIF4A and eIF4G, subunits of the cap–binding complex eIF4F. Consistent with this, we found that eliminating domains in Ded1 that mediate its association with eIF4A or eIF4G increased the Ded1 concentration required for maximal rate acceleration (K1/2Ded1) and, for some mRNAs, also reduced the maximal rate achieved with saturating Ded1 (kmax). Remarkably, the requirements for particular Ded1 interactions with the various components of eIF4F differed substantially for different mRNAs. Thus, Ded1 accelerates 48S assembly by resolving 5′ UTR structures in a manner stimulated by its interactions with eIF4G and eIF4A, supporting the model that Ded1 operates primarily in the context of the quaternary complex eIF4E/eIF4G/eIF4A/Ded1.
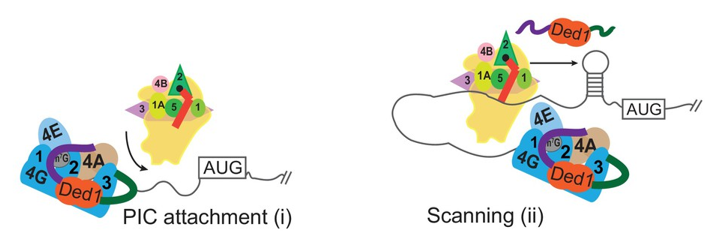
Click image to view.
Figure 1. Different modes of Ded1 function in stimulating PIC attachment to mRNA or subsequent scanning for the start codon
Model depicting how different mRNAs might differ in the extent to which attachment to the mRNA or subsequent scanning of the leader sequence are the rate-limiting steps in 48S PIC assembly. Depending on which step is rate-limiting, the requirements for Ded1, either acting alone or within the eIF4G·eIF4E·eIF4A·Ded1 complex, could be different on different mRNAs.
Functional interplay between RNA helicases Ded1 and Dbp1 in stimulating translation of structured mRNAs in vivo
To illuminate the in vivo function of the Ded1 paralog Dbp1, we conducted ribosome profiling of dbp1Δ and dbp1Δded1 double mutants. The results indicated that Dbp1 functionally cooperates with Ded1 throughout the translatome in stimulating translation of mRNAs with long, structure-prone 5′ UTRs, as the TE reductions in the double mutant generally exceeded those in the ded1 single mutant. For many such mRNAs, Dbp1 largely masks the involvement of Ded1. Importantly, Dbp1 mimics Ded1 in accelerating 48S PIC assembly in the reconstituted system on Ded1–hyperdependent mRNAs containing structured 5′ UTRs. Using the recently developed method of TCP-seq, for genome-wide profiling of 40S subunits to quantify PIC occupancies in 5′ UTRs, we found that 40S subunits tend to accumulate in the 5′ UTRs of mRNAs in the helicase mutants, particularly for mRNAs judged to be Ded1/Dbp1–hyperdependent by conventional 80S profiling, providing direct evidence that Ded1/Dbp1 stimulate scanning through structured 5′ UTRs in vivo to enhance translation. We also uncovered cooperation between these helicases in promoting 43S PIC attachment to a subset of helicase-dependent mRNAs, which exhibit reduced 40S occupancies in 5′ UTRs in the helicase double mutant.
Conserved mRNA–granule component Scd6 utilizes helicase Dhh1 to repress translation initiation and activates Dcp2–mediated mRNA decay in vivo.
Scd6 protein family members are evolutionarily conserved components of mRNA granules. Scd6, and two other proteins with RGG domains, Sbp1 and Npl3, were implicated as translational repressors that act by binding to the RNA2 or RNA3 domains of eIF4G. Scd6 can interact with Dcp2, a catalytic subunit of decapping enzyme, and also with the decapping activator and general translational repressor Dhh1. To provide evidence that Scd6, Sbp1, or Npl3 can function as repressors of particular mRNAs in cells, we tethered each protein, as a fusion to MS2 viral coat protein, to a GFP reporter mRNA containing MS2 coat-protein binding sites in the 3′ UTR region, in wild-type (WT) cells or mutants lacking DCP2 or DHH1. We found that tethering Scd6, but not Sbp1 or Npl3, repressed reporter mRNA abundance in a manner requiring Dcp2, and that it also suppressed reporter mRNA translation via Dhh1. Ribosome profiling and RNA-Seq analysis of scd6Δ and dhh1Δ mutants, and of double mutants also lacking DCP2, indicated that Scd6 cooperates with Dhh1 in both degradation and translational repression of a group of native mRNAs, and that both processes require DCP2, as derepression of both mRNA levels and TEs on deletion of SCD6 or DHH1 was suppressed by dcp2Δ. These last findings lead to the surprising conclusion that translational repression conferred by Scd6 and Dhh1 depend on the decapping enzyme, and possibly generates a pool of uncapped but relatively stable mRNAs of low TEs owing to their inability to bind eIF4F.
Chromatin remodeler Ino80C acts independently of histone variant H2A.Z to evict promoter nucleosomes and stimulate transcription of highly expressed genes.
Previously, we used histone H3 ChIP-Seq analysis to show that that the ATP–dependent chromatin remodeling complexes SWI/SNF and RSC cooperate in achieving wide, nucleosome-depleted regions in the promoters of genes transcriptionally activated by Gcn4, by evicting and repositioning the –1 and +1 promoter nucleosomes, and that defects in this process in mutants lacking these chromatin remodelers (CRs) are associated with reduced transcription. Similar findings were made for the small subset of about 200 most highly transcribed, constitutively expressed genes, suggesting a general cooperation by these CRs in achieving high-level transcription in yeast. The CR Ino80C has been implicated in nucleosome editing by catalyzing replacement of the histone variant H2A.Z (encoded by HTZ1) with canonical H2A. The removal of an H2A.Z:H2B dimer by Ino80C could render the partially disassembled nucleosome more susceptible to eviction; however, an important role for Ino80C in promoter nucleosome eviction had not been reported. By analyzing an ino80Δ mutant lacking the catalytic subunit of Ino80C, we found that this CR functions on par with SWI/SNF in eviction of promoter nucleosomes and transcriptional activation of Gcn4 target genes, and that it plays an even greater role than SWI/SNF at a group of several hundred Ino80C–hyperdependent genes. At Gcn4 target genes, defects in nucleosome eviction are accompanied by reduced promoter occupancies of TATA–binding protein, and hence PIC assembly, and also with reduced transcription. ChIP-seq analysis of Ino80 itself shows that Ino80C is enriched at both Gcn4 target genes and Ino80C–hyperdependent genes.
If Ino80C enhances nucleosome eviction strictly in the course of editing H2A.Z-H2B dimers, then deleting HTZ1 should mimic the effect of deleting INO80 on promoter nucleosome eviction. Moreover, depleting Ino80 should have no effect on nucleosome occupancies in cells lacking HTZ1. At odds with these predictions, the htz1Δ mutation was found to have much smaller effects than ino80Δ on eviction of promoter nucleosomes. Moreover, depleting Ino80 from the nucleus by “anchor-away” impaired histone eviction in the absence of HTZ1. Thus, Ino80C can function similarly to SWI/SNF family members SWI/SNF and RSC in promoting chromatin access independently of nucleosome editing.
Gcn4 binding within coding regions can activate both internal and canonical 5′ promoters in yeast.
We are also interested in determining the role of promoter nucleosome eviction in controlling binding of Gcn4 itself upstream from the promoters of its target genes, and we set out first to define all the binding sites for Gcn4 throughout the genome in wild-type cells. ChIP-seq analysis of Gcn4 binding revealed 546 genomic sites occupied by Gcn4 in starved cells, representing only about 30% of all genomic sequences with significant matches to the consensus Gcn4–binding motif. Analysis of nucleosome occupancy data from MNase-seq analysis revealed that distance of a motif from the nearest nucleosome dyad and its match to the consensus sequence are the major determinants of Gcn4 binding in vivo. Surprisingly, only about 40% of the bound sites are in promoters, and analysis of genome-wide mRNA expression data and ChIP-seq analysis of RNA polymerase II in starvation conditions indicates that only about 60% of such promoter-located Gcn4 occupancy peaks activate transcription, revealing extensive negative control over Gcn4 function. Remarkably, most of the remaining around 300 Gcn4–bound motifs reside within coding sequences (CDS), with about 75 representing the only bound motif in the vicinity of a known Gcn4–induced gene. RNA-seq analysis revealed that many such unconventional Gcn4 occupancy peaks map between divergent antisense and sub-genic sense transcripts induced from within CDS under starvation conditions, and are also located adjacent to starvation-induced TBP (TATA-box binding protein) occupancy peaks detected by ChIP-seq analysis. The findings are consistent with Gcn4 activation of cryptic, bidirectional internal promoters at these genes. Mutational analysis confirmed that Gcn4–bound motifs within CDS can activate both sub-genic and full-length transcripts from the same or adjacent genes, demonstrating that functional Gcn4 binding is not confined to promoters. Our results show that internal promoters can be regulated by a well-defined activator that also functions at conventional 5′ positioned promoters.
Publications
- Rawal Y, Chereji RV, Valabhoju V, Qiu H, Ocampo J, Clark DJ, Hinnebusch AG. Gcn4 binding in coding regions can activate internal and canonical 5' promoters in yeast. Mol Cell 2018;18:30188-30196.
- Zeidan Q, He F, Zhang F, Zhang H, Jacobson A, Hinnebusch AG. Conserved mRNA-granule component Scd6 targets Dhh1 to repress translation initiation and activates Dcp2-mediated mRNA decay in vivo. PLoS Genet 2018;14(12):e1007806.
- Thakur A, Marler L, Hinnebusch AG. A network of eIF2ß interactions with eIF1 and Met-tRNAi promotes accurate start codon selection by the translation preinitiation complex. Nucleic Acids Res 2019;47(5):2574-2593.
- Sen ND, Gupta N, K Archer S, Preiss T, Lorsch JR, Hinnebusch AG. Functional interplay between DEAD-box RNA helicases Ded1 and Dbp1 in preinitiation complex attachment and scanning on structured mRNAs in vivo. Nucleic Acids Res 2019;47(16):8785-8806.
- Gupta N, Lorsch JR, Hinnebusch AG. Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. eLife 2018;7:pii e38892.
Collaborators
- Stuart Archer, PhD, Monash Bioinformatics Platform, Monash University, Australia
- David Clark, PhD, Section on Chromatin and Gene Expression, NICHD, Bethesda, MD
- Chhabi Govind, PhD, Oakland University, Rochester, MI
- Jon Lorsch, PhD, Laboratory on the Mechanism and Regulation of Protein Synthesis, NICHD, Bethesda, MD
- Thomas Preiss, PhD, The John Curtin School of Medical Research, The Australian National University, Canberra, Australia
- Venkatraman Ramakrishnan, PhD, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom
Contact
For more information, email hinnebua@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/hinnebusch.