Cholesterol Homeostasis and Lysosomal Disorders

- Forbes D. Porter, MD, PhD, Head, Section on Molecular Dysmorphology
- Niamh Cawley, PhD, Staff Scientist
- Chris A. Wassif, PhD, Staff Scientist
- Nicole M. Yanjanin, CPNP, Nurse Practitioner
- Antony Cougnoux, PhD, Research Fellow
- Somayeh Hooshmand, PhD, Postdoctoral Fellow
- Kyle Martin, PhD, Postdoctoral Fellow
- Wei-Chia Tseng, PhD, Postdoctoral Fellow
- Insung Kang, PhD, Visiting Fellow
- Anika Prabhu, PhD, Visiting Fellow
- Ana Escauriza, BA, Postbaccalaureate Intramural Research Training Award Fellow
- Mason Fellmeth, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Anna Lyons, BA, Postbaccalaureate Intramural Research Training Award Fellow
- Mojgan Yazdankhah, MS, Postbaccalaureate Intramural Research Training Award Fellow
- Julia Yerger, BA, Postbaccalaureate Intramural Research Training Award Fellow
We study the molecular, biochemical, and cellular processes that underlie genetic disorders resulting from impaired cholesterol homeostasis and lysosomal dysfunction. The disorders include malformation/cognitive impairment syndromes resulting from inborn errors of cholesterol synthesis and neurodegenerative disorders resulting from impaired intracellular cholesterol and lipid transport. Human malformation syndromes attributable to inborn errors of cholesterol synthesis include Smith-Lemli-Opitz syndrome (SLOS), lathosterolosis, desmosterolosis, X-linked dominant chondrodysplasia punctata type 2 (CDPX2), and the CHILD syndrome. Niemann-Pick disease type C (NPC) results from impaired intracellular transport of cholesterol and lipids, leading to neuronal loss. More recently, we focused on Batten disease, which is attributable to mutation of CLN3, the gene that encodes the transmembrane protein battenin. Our research group uses basic, translational, and clinical research approaches with the ultimate goal of developing and testing therapeutic interventions for rare genetic disorders. Our basic research uses neuronal, zebrafish, and mouse models of these genetic disorders to understand the biochemical, molecular, cellular, and developmental processes that underlie the birth defects and clinical problems encountered in affected patients. Our clinical research focuses on translating basic findings to the clinic. Natural history trials of SLOS, CLN3, and NPC1 are ongoing. Our emphasis on both basic and clinical research allows us to integrate laboratory and clinical data in order to improve our understanding of the pathological mechanisms underlying SLOS, CLN3, and NPC, with the goal of improving clinical care of these patients. Therapeutic trials have been conducted for SLOS and NPC1. In collaboration with NCATS (the National Center for Advancing Translational Sciences), our research group has been involved in a multicenter trial of creatine transporter deficiency.
Click image to view.
Figure 1.
Dr. Porter and one of our patients. Neurological exams in children frequently involve 'playing' with the child.
Inborn errors of cholesterol synthesis
Smith-Lemli-Opitz syndrome (SLOS)
SLOS is an autosomal recessive, multiple-malformation syndrome characterized by dysmorphic facial features, cognitive impairment, hypotonia, poor growth, and various structural anomalies of heart, lungs, brain, limbs, gastrointestinal tract, and genitalia. The SLOS phenotype is extremely variable. At the severe end of the phenotypic spectrum, infants often die as result of multiple major malformations, while mild SLOS combines minor physical malformations with behavioral and learning problems. The syndrome is attributable to an inborn error of cholesterol biosynthesis that blocks the conversion of 7-dehydrocholesterol (7-DHC) to cholesterol.
Our laboratory initially cloned the human 3beta-hydroxysterol delta 7-reductase gene (DHCR7) and demonstrated mutations of the gene in SLOS patients. Together with others, we have so far identified over 100 mutations of DHCR7. We also used gene targeting in murine embryonic stem cells to produce several SLOS mouse models, including a null deletion and a hypomorphic point mutation. Mouse pups homozygous for the null mutation (Dhcr7delta3–5/delta3–5) exhibit variable craniofacial anomalies, are growth-retarded, appear weak, and die during the first day of life because they fail to feed. Thus, we were not able to use them to study postnatal brain development, myelination, or behavior or to test therapeutic interventions. For this reason, we developed a missense allele (Dhcr7T93M). The T93M mutation is the second most common mutation found in SLOS patients. Dhcr7T93M/T93M and Dhcr7T93M/delta3–5 mice are viable and demonstrate SLOS with a gradient of biochemical severity (Dhcr7delta3–5/delta3–5 greater than Dhcr7T93M/delta3–5 greater than Dhcr793M/T93M). We used Dhcr7T93M/delta3–5 mice to test the efficacy of therapeutic interventions on tissue sterol profiles. As expected, dietary cholesterol therapy improved the sterol composition in peripheral tissues but not in the central nervous system. Treatment of mice with the statin simvastatin improved the biochemical defect in both peripheral and central nervous system tissue, suggesting that simvastatin therapy may be used to treat some of the behavioral and learning problems in children with SLOS. Most recently, we developed a zebrafish model for SLOS that will allow us to study the impact of aberrant cholesterol synthesis on behavior. Characterization of induced pluripotent stem cells from SLOS patients demonstrated a defect in neurogenesis, which results from inhibition of Wnt signaling owing to a toxic effect of 7-DHC.
As part of our clinical studies on SLOS, we identified a novel oxysterol, 27-hydroxy-7-dehydrocholesterol (27-7DHC), derived from 7-DHC in SLOS patients. We therefore investigated whether 27-7DHC contributes to the pathology of SLOS and found a strong negative correlation between plasma 27-7DHC and cholesterol levels in these patients. In addition, previous work showed that low cholesterol levels impair hedgehog signaling (a signaling pathway required for proper cell differentiation). Therefore, we hypothesized that increased 27-7DHC levels would have detrimental effects during development as a result of suppression of cholesterol levels. To test our hypothesis, we produced SLOS mice (Dhcr7delta3–5/delta3–5) expressing a CYP27 (sterol 27-hydroxylase) transgene. CYP27Tg mice display increased CYP27 expression and elevated 27-hydroxycholesterol levels but normal cholesterol levels. While Dhcr7delta3–5/delta3–5 mice are growth-retarded, exhibit a low incidence of cleft palate (9%), and die during the first day of life, Dhcr7delta3–5/delta3–5:CYP27Tg embryos are stillborn and have multiple malformations, including growth retardation, micrognathia, cleft palate (77%), lingual and dental hypoplasia, ankyloglossia, umbilical hernia, cardiac defects, cloacae, curled tails, and limb defects; we also observed autopod defects (polydactyly, syndactyly, and oligodactyly) in 77% of the mice. Consistent with our hypothesis, sterol levels were halved in the liver and 20-fold lower in the brain tissue of Dhcr7delta3–5/delta3–5:CYP27Tg than in Dhcr7delta3–5/delta3–5 embryos. The fact that 27-7DHC plays a role in SLOS may explain some of the phenotypic variability and may lead to development of a therapeutic intervention. The project is a good example of the benefits of integrating clinical and basic science to both understand the pathology of SLOS and develop potential therapeutic interventions. We are currently investigating the pathological role of other 7-DHC–derived oxysterols, such as DHCEO (3beta,5alpha-dihydroxy-cholest-7-en-6-one).
Development of patient-derived induced pluripotent stem cells has given us insight into fundamental mechanisms that impair neuronal development in SLOS.
We are conducting a longitudinal Natural History trial. Given that SLOS patients have a cholesterol deficiency, they may be treated with dietary cholesterol supplementation. To date, we have evaluated over 100 SLOS patients.
One reason for studying rare genetic disorders is to gain insight into more common disorders. Most patients with SLOS exhibit autistic characteristics. We are currently collaborating with other NIH and extramural groups to further evaluate this finding.
Lathosterolosis and desmosterolosis
Lathosterol 5-desaturase catalyzes the conversion of lathosterol to 7-dehydrocholesterol, representing the enzymatic step immediately preceding the defect in SLOS. Thus, to gain a deeper understanding of the roles of reduced cholesterol versus elevated 7-dehydrocholesterol levels in SLOS, we disrupted the mouse lathosterol 5-desaturase gene (Sc5d) by using targeted homologous recombination in embryonic stem cells. Sc5d–/– pups are stillborn, present with micrognathia and cleft palate, and exhibit limb-patterning defects. Many of the malformations in the mutant mice resemble malformations in SLOS and are consistent with impaired hedgehog signaling during development. Biochemically, the mice exhibit markedly elevated lathosterol levels and reduced cholesterol levels in serum and tissue.
Desmosterolosis is another inborn error of cholesterol synthesis that resembles SLOS. It results from a mutation in the 3beta-hydroxysterol delta 24-reductase gene (DHCR24). DHCR24 catalyzes the reduction of desmosterol to cholesterol. We disrupted the mouse Dhcr24 gene with targeted homologous recombination in embryonic stem cells. Surprisingly, although most Dhcr24 mutant mice die at birth, the pups are phenotypically normal.
Niemann-Pick disease type C1
Niemann-Pick disease type C1 (NPC1) is a neurodegenerative disorder that results in ataxia and dementia. In view of the dementia, it has been referred to as childhood Alzheimer's disease. The disorder is caused by a defect in intracellular lipid and cholesterol transport. Initially, as part of a Bench-to-Bedside award, we began a clinical protocol to identify and characterize biomarkers that could be used in a subsequent therapeutic trial. The project also received support from the Ara Parseghian Medical Research Foundation and Dana’s Angels Research Trust. We have enrolled over 100 NPC1 patients in a longitudinal Natural History trial. The goals of the trial are to identify (1) a blood-based diagnostic/screening test, (2) biomarkers that can be used as tools to facilitate development and implementation of therapeutic trials, and (3) clinical symptoms/signs that may be used as efficacy outcome measures in a therapeutic trial.
Click image to view.
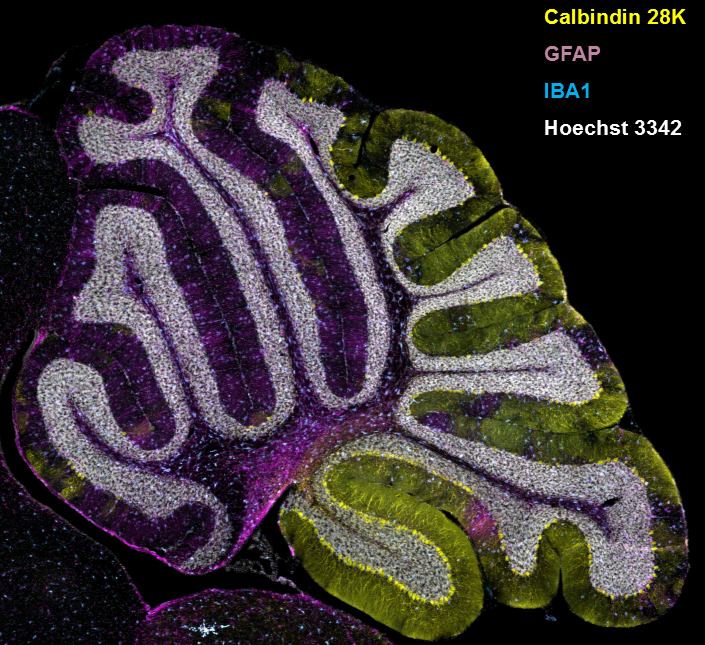
Figure 2. Gliosis in NPC1 mouse cerebellum
Immuno-staining of a sagittal section from the cerebellum of an NPC1–mutant mouse. Cerebellar Purkinje neurons are stained for calbindin 28K, and the expected loss of anterior Purkinje neurons is readily apparent. Expression of GFAP and IBA1 are used to detect astrogliosis and microgliosis, respectively. Nuclei are stained with Hoechst 3342.
Currently, the average time from first symptom to diagnosis, the 'diagnostic delay,' in our cohort of NPC patients is on the order of four to five years. In collaboration with Daniel Ory, we found elevated levels of nonenzymatically produced oxysterols in NPC1 patients. As well as a potential biomarker that may be used to follow therapeutic interventions, testing for oxysterols or bile acid derivatives has now become a standard method of diagnosis.
In addition to our Natural History study, we completed a randomized, placebo-controlled, cross-over trial to investigate the safety and efficacy of N-acetyl cysteine (NAC) in NPC1. The goal was to determine whether NAC treatment would reduce oxidative stress and subsequently lower levels of the nonenzymatically produced oxysterols. We also tested the safety and efficacy of the histone deacetylase (HDAC) inhibitor vorinostat in adult NPC1 patients. In collaboration with the Therapeutics of Rare and Neglected Disease Program of NCATS, we completed a phase 1/2a therapeutic trial of lumbar intrathecal cyclodextrin (VTS-270, adrabetadex) therapy in NPC1. We have now transitioned to a multicenter, multinational phase 2b/3, which is being evaluated. We also initiated a study to evaluate the safety and efficacy of combined intrathecal and intravenous cyclodextrin.
To complement the clinical work, we have begun to apply molecular and proteomic approaches to both mouse and human biomaterials in order to identify biological pathways disrupted in NPC1. We identified several blood and CSF (cerebral spinal fluid) proteins and are in the process of validating the biomarkers as potential outcome measures to be used as tools in the development of therapeutic interventions.
Click image to view.
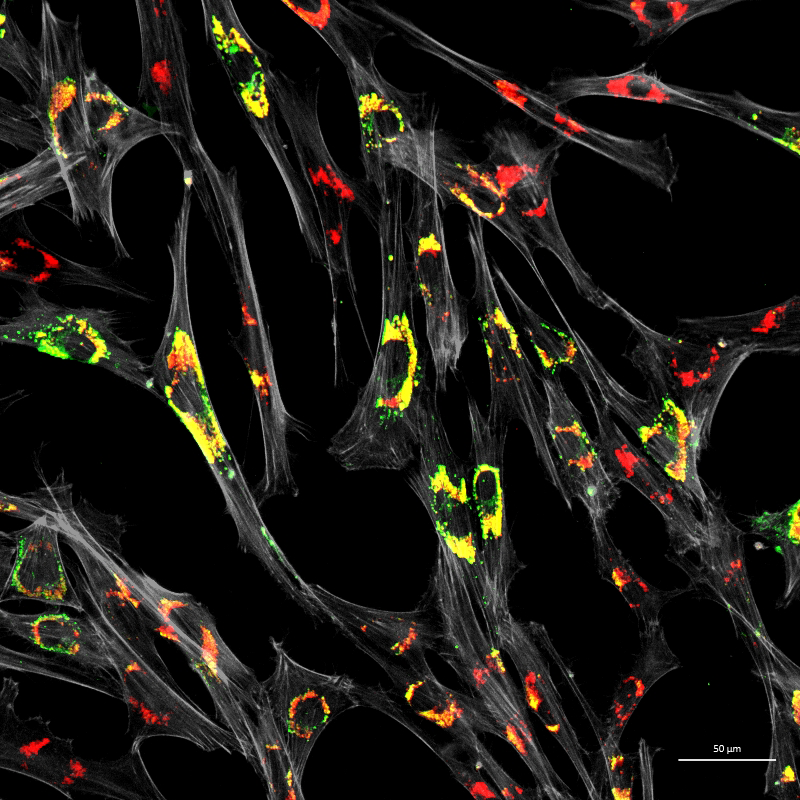
Figure 3. Accumulation of unesterified cholesterol in NPC1 patient fibroblasts
Human NPC1 fibroblasts were immuno-stained for Lamp1 (green) and stained with filipin (red); filipin stains unesterified cholesterol, which accumulates in the Lamp1–positive endolysosomal compartment. Cell structure was outlined by immuno-staining for actin (gray).
Creatine transport deficiency and CLN3 Disease
Recently, we initiated natural history protocols to study children with creatine transport deficiency (CTD) and CLN3 disease (juvenile Batten disease). CTD is an X-linked disorder arising from mutation of SLC6A8 (which encodes Solute Carrier Family 6 Member 8, a protein called sodium- and chloride-dependent creatine transporter 1). Individuals with CTD manifest significant developmental delay and have frequent seizures. The work on CTD is a multicenter trial being conducted in collaboration with NCATS and Lumos Pharma. Our goal is to obtain detailed natural history data, establish a biorepository, find biomarkers, and identify potential clinical outcome measures in preparation for a therapeutic trial.
CLN3 disease (juvenile Batten disease) is an autosomal recessive, progressive neurodegeneration arising from mutation of CLN3, the gene encoding the lysosomal/endosomal protein battenin. The function of the battenin is not known, but its absence leads to a lysosomal storage disorder. Children with CLN3 disease typically first lose vision, followed by progressive cognitive and motor impairment. Similar to the other disorders that we study, our goal is to conduct a natural history study in order to facilitate studies designed to understand the pathology underlying these disorders as well as development of therapeutic interventions.
Additional Funding
- Bench to Bedside award: Investigations of Juvenile Neuronal Ceroid Lipofuscinosis (CLN3)
- U01HD0990845: Intravenous delivery of 2-hydroxypropyl-beta-cyclodextrin for treatment of Niemann-Pick C disease
- Ara Parshegian Medical Research Fund, University of Notre Dame
- Together Strong NPC Foundation
- Dana's Angels Research Trust
Publications
- Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med 2010;2:56ra81.
- Francis KR, Ton AN, Xin Y, O'Halloran PE, Wassif CA, Malik N, Williams IM, Cluzeau CV, Trivedi NS, Pavan WJ, Cho W, Westphal H, Porter FD. Modeling Smith-Lemli-Opitz syndrome with induced pluripotent stem cells reveals a causal role for Wnt/beta-catenin defects in neuronal cholesterol synthesis phenotypes. Nat Med 2016;22:388-396.
- Tseng WC, Loeb HE, Pei W, Tsai-Morris CH, Xu L, Cluzeau CV, Wassif CA, Feldman B, Burgess SM, Pavan WJ, Porter FD. Modeling Niemann-Pick disease type C1 in zebrafish: a robust platform for in vivo screening of candidate therapeutic compounds. Dis Model Mech 2018;11:dmm034165.
- Cougnoux A, Drummond RA, Collar AL, Iben JR, Salman A, Westgarth H, Wassif CA, Cawley NX, Farhat NY, Ozato K, Lionakis MS, Porter FD. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum Mol Genet 2018;27:2076-2089.
- Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener L, Rao R, Soldatos A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD. Intrathecal 2-hydroxypropyl-ß-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: an ad-hoc analysis of a non-randomized, open-label, phase 1/2 trial. Lancet 2017;390(10104):1758-1768.
Collaborators
- Daniel Ory, MD, Washington University in St. Louis, St. Louis, MO
- William Pavan, PhD, Genetic Disease Research Branch, NHGRI, Bethesda, MD
Contact
For more information, email fdporter@helix.nih.gov or visit https://irp.nih.gov/pi/forbes-porter.