Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Lisa Zhang, PhD, Staff Scientist
- Chi-Jiunn Pan, BS, Senior Research Assistant
- Jun Ho Cho, PhD, Visiting Fellow
- Goo Young Kim, PhD, Visiting Fellow
- Joon Hyun Kwon, PhD, Visiting Fellow
- Javier Anduaga, BS, Technical Intramural Research Training Award Fellow
- Kurt Berckmueller, BS, Predoctoral Intramural Research Training Award Fellow
- Vamsi Reddy, BS, Predoctoral Intramural Research Training Award Fellow
- Young Mok Lee, PhD, Guest Researcher
- Brian C. Mansfield, PhD, Guest Researcher
We conduct research to delineate the pathophysiology of glycogen storage disease type I (GSD-I) and glucose-6-phosphatase-beta (G6Pase-beta or G6PC3) deficiency (GSD-Irs) and to develop novel therapies for these disorders. GSD-I consists of two subtypes, GSD-Ia, deficient in G6Pase-alpha (or G6PC), and GSD-Ib, deficient in the glucose-6-phosphate (G6P) transporter (G6PT or SLC37A4). A third disease, G6Pase-beta deficiency, also known as severe congenital neutropenia syndrome type 4, is not a glycogen storage disease but biochemically a GSD-I–related syndrome (GSD-Irs). G6Pase-alpha and G6Pase-beta are endoplasmic reticulum (ER)–bound G6P hydrolases, with active sites lying inside the lumen, which depend upon G6PT to translocate G6P from the cytoplasm into the ER lumen. The G6PT/G6Pase-alpha complex maintains interprandial glucose homeostasis, while the G6PT/G6Pase-beta complex maintains energy homeostasis and functionality of neutrophil and macrophages. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis not shared by GSD-Irs. GSD-Ib and GSD-Irs patients manifest a common myeloid phenotype of neutropenia and myeloid dysfunction not shared by GSD-Ia. Neutrophils express the G6PT/G6Pase-beta complex, and inactivation of G6PT or G6Pase-beta leads to the enhanced neutrophil apoptosis that underlies neutropenia in GSD-Ib and GSD-Irs. The G6PT/G6Pase-beta complex is also essential for energy homeostasis in neutrophils. A deficiency in either G6PT or G6Pase-beta prevents recycling of glucose from the ER to the cytoplasm, leading to neutrophil dysfunction. There is no cure for GSD-Ia, GSD-Ib, or GSD-Irs. Animal models of the three disorders are available and are being exploited to both delineate the diseases more precisely and to develop new treatment approaches, including gene therapy.
Functional analysis of mutations in GSD-Irs
The enzyme G6Pase-beta is embedded in the ER membrane and catalyzes the hydrolysis of G6P to glucose and phosphate. To date, 33 distinct G6Pase-beta mutations have been identified in GSD-Irs patients, but only the p.R253H and p.G260R missense mutations have been characterized functionally for pathogenicity. We functionally characterized 16 of the 19 known missense mutations, using a sensitive assay based on a recombinant adenoviral vector–mediated expression system to demonstrate pathogenicity. Twelve missense mutations completely abolish G6Pase-beta enzymatic activity, while the p.M116V, p.T118R, p.S139I, and p.R189Q mutations retain 1.1%, 1.3%, 49%, and 45%, respectively, of wild-type G6Pase-beta activity. A database of residual enzymatic activity retained by the G6Pase-beta mutations will serve as a reference for evaluating genotype-phenotype relationships.
Mechanisms preventing hepatocellular adenoma/carcinoma in GSD-Ia mice receiving gene therapy
The hallmarks of GSD-Ia are impaired glucose homeostasis and long-term risk of hepatocellular adenoma/hepatocellular carcinoma (HCA/HCC). The predominant subtypes of HCA in GSD-Ia are inflammatory HCA (IHCA, 52%) and β-catenin–mutated HCA (bHCA, 28%). We previously showed that AAV (adeno-associated virus) mice that express more than five units of hepatic G6Pase-α activity maintain glucose homeostasis, demonstrate no HCA/HCC, and live under CR (caloric restriction). We recently showed that 75% of rAAV–treated mice expressing 1.5-4.9 units of hepatic G6Pase-α activity (AAV-low-NT mice) exhibit a similar phenotype. In view of the similarities between AAV-NT (NT, non-tumor) mice and animals living under CR, we examined pathways involved in CR for insights into mechanisms underlying the absence of HCA/HCC in AAV/AAV-low-NT (AAV-NT) mice. The CR mediators 5′ adenosine monophosphate–activated protein kinase (AMPK) and sirtuin-1 (SIRT1), which share activators, actions, and targets, were activated in AAV-NT mice. AMPK elicits anti-inflammatory action by inhibiting phosphorylation and activation of STAT3, a transcription factor that promotes cancer-causing inflammation. SIRT1 represses NFκB activity via deacetylates of NFκB-p65, a transcription factor that promotes inflammation-associated cancer. The signaling by STAT3 and NF-κB is highly interconnected. Together they regulate several genes involved in tumor proliferation, survival, and invasion. In AAV-NT mice, hepatic levels of active p-STAT3-Y705 and ac-NFκB-p65-K310 were reduced. SIRT1 also inhibits cancer metastasis. We showed that the expression of mesenchymal markers, STAT3 targets, NFκB targets, and β-catenin targets, which all promote tumorigenesis, were also reduced. AAV-NT mice also expressed increased levels of E-cadherin, FGF21, and the tumor suppressor β-klotho. Importantly, treating the mice with the SIRT1 inhibitor EX-527 markedly reversed many of the anti-tumorigenic pathways. In summary, activation of hepatic AMPK/SIRT1 and FGF21/β-klotho signaling combined with down-regulation of STAT3 and NFκB signaling underlie the absence of HCA/HCC in AAV-NT mice.
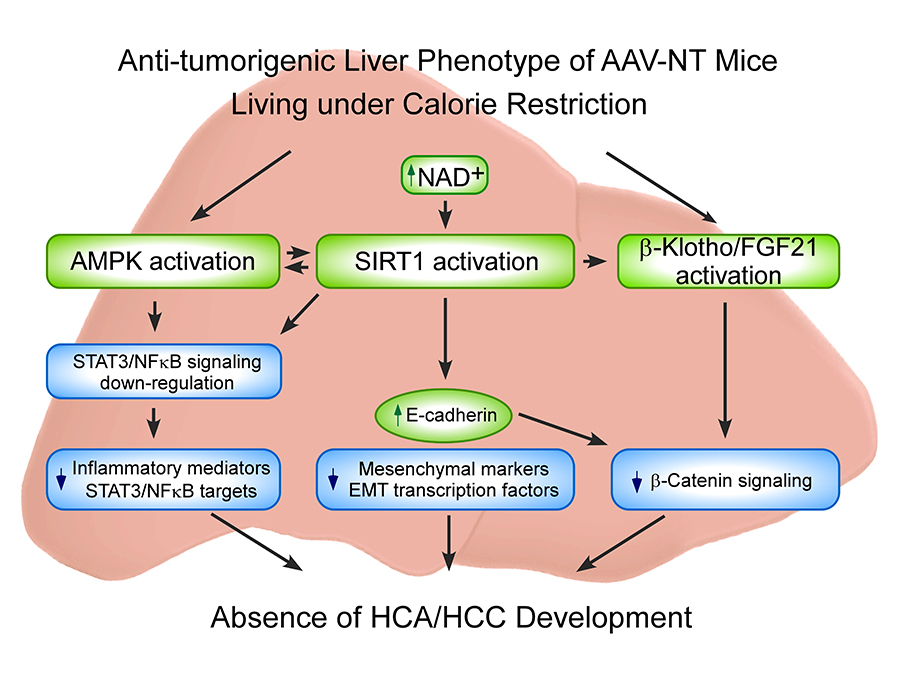
Click image to enlarge.
Proposed mechanisms that prevent HCA/HCC in G6pc–/– mice receiving gene therapy
Several signaling pathways contribute to the anti-tumorigenic liver phenotype of the AAV-NT mice. The increases in total AMPK and p-AMPK-T172 activate AMPK, which suppresses phosphorylation and activation of STAT3 signaling. The increase in hepatic NAD+ levels activates SIRT1, which suppresses NFκB signaling via deacetylation of the p65 subunit of NFκB. The down-regulation of hepatic STAT3/NFκB signaling leads to reduced expression of pro-inflammatory mediators and STAT3/NFκB targets. SIRT1 activation also negatively regulates tumor metastasis by increasing the expression of E-cadherin and reducing the expression of mesenchymal markers and the epithelial-mesenchymal transition transcription factors. The increase in E-cadherin also inhibits β-catenin signaling which plays a key role in the pathogenesis of HCC. CR also activates FGF21/β-klotho signaling, which decreases the expression of β-catenin targets and prevents hepatocarcinogenesis.
Molecular mechanisms underlying the improved metabolic phenotype in GSD-Ia mice receiving gene therapy
We showed that gene therapy mediated by rAAV8-G6PC, a recombinant adeno-associated virus pseudotype 2/8 (rAAV8) vector expressing human G6Pase-alpha directed by the human G6PC promoter/enhancer, normalizes blood glucose homeostasis in G6pc knockout (G6pc–/–) mice for 70-90 weeks. The treated G6pc–/– mice (AAV mice) expressing 5 units or more of hepatic G6Pase-alpha activity (3% or more of normal hepatic G6Pase-alpha activity) maintain glucose homeostasis, show no evidence of hepatocellular adenoma (HCA)/carcinoma (HCC), and are protected against age-related insulin resistance or obesity. We then undertook studies to delineate the molecular mechanisms underlying this beneficial metabolic phenotype. Studies showed that mice over-expressing hepatic carbohydrate-responsive element–binding protein (ChREBP) exhibit improved glucose and lipid metabolism, resulting from activation of the protein kinase Akt and increased expression of stearoyl-CoA desaturase 1 (SCD1), which converts saturated fatty acids into the beneficial mono-unsaturated fatty acids. In AAV mice, elevated concentrations of hepatic G6P stimulate hepatic ChREBP signaling, leading to increased SCD1 and activation of the protein kinases p-Akt-S473 and p-Akt-T308, consistent with activation of ChREBP signaling, as one mechanism underlying the beneficial phenotype.
We further showed that the AAV mice mimic animals living under CR. The major downstream modulators of CR are AMPK, a master regulator of energy homeostasis, and SIRT1, an NAD+–dependent deacetylase. We showed that hepatic NAD+ concentrations in the AAV mice are higher than in age-matched control mice, consistent with SIRT1 activation. The AAV mice also exhibit increased protein levels of AMPK and active p-AMPK-T172. Given that aging is associated with mitochondrial dysfunction, protection of mitochondria may also contribute to the observed phenotype. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a master regulator of energy metabolism and mitochondrial biogenesis and plays key roles in the maintenance of mitochondrial integrity, biogenesis, and function. We showed that the expression of PGC-1α, which can be activated both by AMPK–mediated phosphorylation and SIRT1–mediated deacetylation, is up-regulated in the AAV mice. Moreover, protein levels of complex I to V of the mitochondrial electron transport chain are up-regulated in AAV mice. Taken together, the underlying mechanisms responsible for the beneficial metabolic phenotype correlate with activation of ChREBP and AMPK/SIRT1/PGC-1α signaling pathways in the livers of AAV mice.
Molecular mechanisms underlying HCA and HCC in murine GSD-Ia
The most severe complications in GSD-Ia are the development of HCA and HCC of unknown etiology. Global knock-out G6pc–/– mice die early, well before HCA/HCC can develop, making studies of HCA/HCC mechanisms difficult. We therefore generated liver-specific G6pc knock-out mice (L-G6pc–/–) that survive to adulthood and develop HCA. Using L-G6pc–/– mice, we investigated the underlying mechanisms of HCA/HCC in GSD-Ia. Studies have shown that autophagy-deficient mice develop HCA and we looked to this as a guide. We hypothesized that defective autophagy may underlie HCA/HCC development in GSD-Ia.
Autophagy can be regulated directly by SIRT1 through deacetylation of autophagy-related (ATG) proteins and indirectly though deacetylation and activation of FoxO factors, which transactivate autophagy genes. SIRT1 activity can be activated by the cofactor NAD+, but levels of NAD+ were unchanged in L-G6pc–/– livers. The expression of SIRT1 can be suppressed by lipogenic factors such as ChREBP and peroxisome proliferator-activated receptor gamma (PPAR-γ) and stimulated by FoxO1 (a transcription factor that plays important roles in regulation of gluconeogenesis and glycogenolysis by insulin signaling) and PPAR-α in response to nutrient starvation. In the G6pc–/– liver, activation of ChREBP signaling was associated with increased lipogenesis and suppressed PPAR-α expression, leading to a marked increase in hepatic steatosis and elevated PPAR-γ expression. The net outcome was a lower expression of SIRT1 in G6pc–/– livers than in controls. In addition, protein levels of FoxO3a were lower in G6pc–/– livers. Collectively, reduced SIRT1-FoxO signaling appears to contribute to impaired autophagy in G6pc–/– livers. The G6Pase-α–deficient liver also displays mitochondrial dysfunction characterized by a reduction in oxidative phosphorylation, lower overall mitochondrial numbers, and a decrease in functional mitochondria. The mechanism underlying mitochondrial dysfunction arises from down-regulation of hepatic SIRT1-PGC-1α signaling pathway.
We further showed that the underlying mechanisms responsible for HCA/HCC formation in GSD-Ia include accumulation of polyubiquitin-binding protein (p62) aggregates, which promotes tumorigenesis, marked mitochondrial, and oxidative DNA damage. Importantly, we showed that restoration of hepatic G6Pase-α expression by rAAV-G6PC–mediated gene transfer normalizes autophagy deficiency and restores SIRT1-FoxO signaling in L-G6pc–/– mice. Taken together, our study provides the underlying mechanisms for HCA/HCC in GSD-Ia and also suggests that correction of defective autophagy by gene therapy or pharmacological intervention will prevent or slow the chronic development of HCA/HCC in human GSD-Ia patients.
Liver-directed gene therapy for murine GSD-Ib
We showed that systemic administration of rAAV-CBA-G6PT, a rAAV8 vector expressing human G6PT directed by the chicken b-actin (CBA) promoter/CMV (cytomegalovirus) enhancer, delivered the G6pt transgene to the liver and normalized metabolic abnormalities in murine GSD-Ib. However, the five transduced GSD-Ib mice that lived to age 52–70 weeks expressed less than 4% of wild-type hepatic G6PT activity, and two mice developed HCA, with one undergoing malignant transformation. Studies have shown that the choice of transgene promoter not only affects targeting efficiency and tissue-specific expression but also the level of immune response or tolerance to the therapy. We therefore examined the safety and efficacy of rAAV8-G6PT, a rAAV8 vector expressing human G6PT directed by the tissue-specific human G6PC promoter/enhancer. Among the fifteen 60–78 week-old rAAV–treated G6pt–/– mice expressing 2–62% of wild-type hepatic G6PT activity, only one mouse expressing 6% of normal hepatic G6PT activity developed HCA. The rAAV–treated mice, including the HCA–bearing mouse, displayed normal hepatic fat storage, normal glucose tolerance profiles, and maintained normoglycemia over a 24-hour fast. The rAAV–treated mice also exhibit a leaner phenotype and are protected against age-related insulin resistance. We further showed that activation of hepatic ChREBP signaling, which improves glucose tolerance and insulin sensitivity, is one mechanism that protects the rAAV-GPE-G6PT–treated G6pt–/– mice against age-related obesity and insulin resistance.
Additional Funding
- The Children's Fund for Glycogen Storage Disease Research, 2015
- Dimension Therapeutics (Cambridge, MA) under a Cooperative Research and Development Agreement (CRADA)
Publications
- Lin SR, Pan CJ, Mansfield BC, Chou JY. Functional analysis of mutations in a severe congenital neutropenia syndrome caused by glucose-6-phosphatase-beta deficiency. Mol Genet Metab 2015;114:41-45.
- Kim GY, Lee YM, Cho JH, Pa CJ, Jun HS, Springer DA, Mansfield BC, Chou JY. Mice expressing reduced levels of hepatic glucose-6-phosphatase-a activity do not develop age-related insulin resistance and obesity. Hum Mol Genet 2015;24:5115-5125.
- Chou JY, Jun HS, Mansfield BC. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J Inherit Metab Dis 2015;38:511-519.
- Lee YM, Kim GY, Pan CJ, Mansfield BC, Chou JY. Minimal hepatic glucose-6-phosphatase-a activity required to sustain survival and prevent hepatocellular adenoma formation in murine glycogen storage disease type Ia. Mol Genet Metab Rep 2015;3:28-32.
Collaborators
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genova, Italy
- Luigi Varesio, PhD, Istituto Giannina Gaslini, Genova, Italy
- David A. Weinstein, MD, University of Florida College of Medicine, Gainesville, FL
Contact
For more information, email chou@helix.nih.gov or visit https://irp.nih.gov/pi/janice-chou.