Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Mahesh Bachu, PhD, Visiting Fellow
- Chao Chen, PhD, Visiting Fellow
- Vishal Nehru, PhD, Visiting Fellow
- Ryota Oda, PhD, Visiting Fellow
- Sumihito Togi, PhD, Visiting Fellow
- Keith Sakata, BS, Postbaccalaureate Intramural Research Training Award Fellow
Macrophages and related cells recognize incoming pathogens and produce cytokines, such as interferons (IFNs) and IL-1/IL-6/TNF-alpha. While IFNs impart anti-viral and anti-microbial protection to the host, the latter cytokines are associated with inflammatory responses. IFNs are produced upon activation of the IRF (interferon regulatory factor) family of transcription factors, while inflammatory cytokines are produced by the activation of the transcription factor NFκB. Our goal is to study the molecular pathways that direct the development and function of macrophages and other myeloid cells. To this end, we focus on the role of IRF8 in innate immunity. IRF8, a member of the IRF family, is expressed in macrophages, dendritic cells (DCs), and microglia at high levels, and is required for the production of both type I and type II IFNs. IRF8 is essential for mounting the first line of defense against various invading pathogens prior to the initiation of antigen-specific adaptive immune responses.
Transcriptionally active genes are embedded in chromatin that is dynamically exchanged, whereas silenced genes are surrounded by more stable chromatin. The chromatin environment contributes to the epigenetic states of given cells and influences transcriptional processes. We have long been working on BRD4, a bromodomain protein that binds to acetylated histones and promotes active transcription. BRD4 is involved in the dynamic chromatin exchange that takes place in highly transcribed genes. The exchange requires a special histone called H3.3. As a result of the association with transcription, H3.3 is implicated in epigenetic control of gene expression patterns. Our goal is to elucidate the activity of BRD4 and histone H3.3 in innate immunity.
IRF8 is a master regulator of autophagy in macrophages and restricts Listeria monocytogenes replication.
Autophagy is a highly conserved catabolic process to degrade misfolded self-proteins and damaged organelles. Autophagy is essential for embryonic development and occurs in a wide variety of adult cells. Macrophages and DCs use this mechanism to fortify their ability to eliminate invading pathogens. Autophagy plays a critical role in innate resistance to various bacteria, including Mycobacterium tuberculosis and Salmonella. Autophagy is activated by many stresses, including IFN-gamma–toll like receptor stimulation and bacterial infection. In addition, starvation and macrophage colony-stimulating factor (M-CSF) stimulation trigger autophagy in macrophages. Our initial gene profiling analysis in DCs pointed to the role of IRF8 in autophagy gene activation. Subsequent analysis confirmed that 17 autophagy genes are downregulated in Irf8–/– macrophages. The IRF8–regulated autophagy genes include those active at all steps of autophagy, from early signaling, autophagosome formation, lysosomal fusion, and degradation of captured materials. Using the ChIP-PCR technique, we confirmed that IRF8 binds to the upstream elements of many autophagy genes. Interestingly IRF8 was upregulated following all stresses mentioned above. As a result, Irf8–/– macrophages were defective in autophagic activity and accumulated the ubiquitin-binding protein SQSTM1 in the cytoplasm, forming harmful aggregates. We further showed that Irf8–/– macrophages were highly susceptible to Listeria monocytogenes infection. When infected with Listeria, many autophagy genes in wild-type macrophages were transcriptionally activated, leading to autophagic capturing and degradation of the bacteria. In contrast, these processes were absent from Irf8–/– macrophages, leading to uncontrolled bacterial growth (Figure 1). Thus IRF8 serves as a master regulator of autophagy in macrophages and promotes innate resistance to infection and macrophage survival.
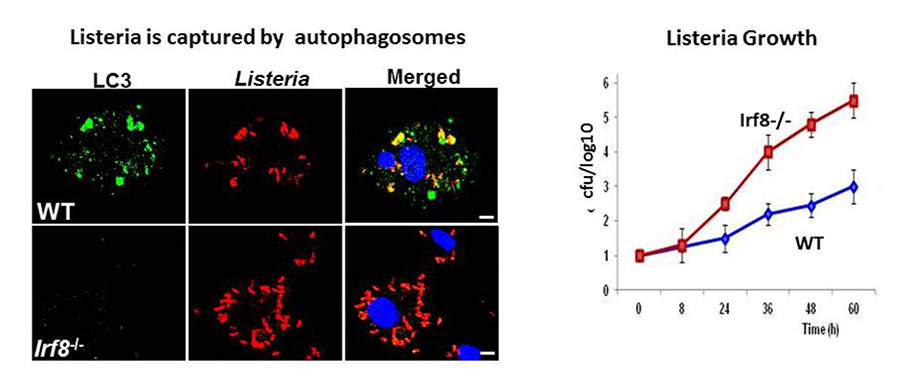
Click image to enlarge.
Figure 1. IRF8 is a master regulator of autophagy in macrophages.
Left: Wild-type (WT) and Irf8–/– macrophages were infected with Listeria monocytogenes for 3 days, and localization of the Listeria antigens (red) and activated autophagy component LC3 (green) was examined by immunostaining. Blue color represents DNA (nucleus) by DAPI stain. Irf8–/– cells had fewer LC3 punctae and contained many more Listeria than wild-type cells.
Right: Listeria replication was tested by the colony-forming unit (cfu) assay. Data show that the bacteria grew exponentially in Irf8–/– macrophages, while bacterial growth was restricted in their wild-type counterparts.
We also investigated innate resistance to Ebola virus (EBOV) infection and found IRF8–dependent type I IFN signaling to be important. EBOV causes a severe hemorrhagic disease with high fatality. In collaboration with scientists at the United States Army Medical Research Institute, we showed that virus-like particles (VLPs) from EBOV stimulate toll-like receptor (TLR) signaling and then the IFN (JAK-STAT) pathway in vitro. We then showed that VLPs protect mice from lethal EBOV infection when given before and after EBOV infection. However, Ifnar knockout (KO) mice died even after VLP treatment, while wild-type mice were fully protected. VLPs caused accelerated induction of interferon-stimulated genes (ISGs) in wild-type but not in Ifnar KO mice, a likely mechanism of protection. Moreover, VLP protection required IRF8, which boosted IFN feedback signaling.
Epigenetic states of the IRF8 locus in human cells
IRF8 binds to two motifs in macrophages and developing myeloid cells. In collaboration with others, we studied the genome-wide distribution of IRF8. The studies highlighted two widely distributed DNA motifs to which IRF8 bound: ETS/IRF composite motif (GGAA and GAAA and similar) and the ISRE–like motif, the GAAA repeat. For example, IRF8 bound to a distal enhancer of the Klf4 gene, through which it promoted myeloid cell differentiation. We found that IRF8 binding is enriched with the composite sites in developing monocytes while IRF8 binding to ISRE–like sites rose upon stimulation in bone marrow (BM) macrophages. The studies serve as a steppingstone for our future work to analyze chromatin environment important for macrophage innate immunity. To assess epigenetic features of the IRF family genes, we mined publicly available data on the post-translational histone modification marks reported in various human cells, which are deposited in the ENCODE (Encyclopedia of DNA Elements) repository at the University of California Santa Cruz (https://genome.ucsc.edu/ENCODE). We found that IRF family genes can be classified into several distinct groups according to the pattern of histone modifications. Acetylation of H3 and H4 and methylation of H3 at K4, K9, and K27 were major features assisting our classification (Figure 2). Interestingly, these chromatin features matched their functional properties and expression patterns. The analysis strengthened the notion that chromatin regulators are unrecognized players in IRF–family activity.
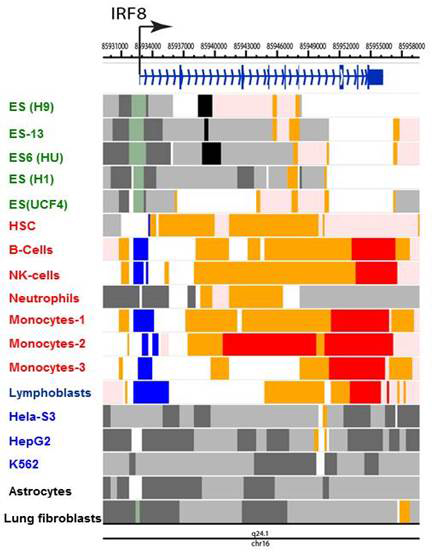
Click image to enlarge.
Figure 2. Epigenetic states of the IRF8 locus in human cells
Epigenome landscape of hIRF8 gene in ES (gray), ex vivo immune cells (red) and cell lines (black). Data were obtained from the ENCODE program and reorganized to visualize indicated chromatin states, using the Washington University Epigenome Browser (http://epigenomegateway.wustl.edu/browser). Cells examined were embryonic stem (ES) cells (green), primary immune cells (red), and cancer cell lines (blue). Chromatin states were assessed by the indicating modifications: active TSS (H3K3me3, H3K4me2, H3K27Ac, H3K9Ac); bivalent promoter (H3K4me3, H3K27me3); strong promoter (H3K36me3, H4K20me1, H3K79me2); weak or no transcription (H3K36me3, H4K20me1, H3K79me2); enhancer (H3K4me1, H3K27Ac, H3K9Ac); heterochromatin (H3K9me3); polycomb repression (H3K27me3).
BRD4 supports elongation of the eRNA and the coding RNA.
BRD4 has two bromodomains through which it binds to the acetylated histones H3 and H4. BRD4 also interacts with P-TEFb, the elongation factor composed of Cyclin T and CDK9. The interaction relieves the 5′-paused RNA polymerase II (Pol II) to trigger elongation. Recent studies from various laboratories showed that BRD4 promotes cancer cell growth, including several forms of leukemia, primarily by promoting c-Myc expression. Interestingly, pharmacological inhibitors for bromodomains in BET (bromodomain and extra-terminal) proteins are found to be effective anti-leukemia drugs in animal models. To further study the extent of BRD4 involvement in elongation, we performed RNA-seq and ChIP-seq analyses of chromatin-bound RNA in quiescent and serum-stimulated fibroblasts. We were particularly interested in the enhancer regions that carry epigenetic histone marks, such as H3K4me1 and H3K27ac, thereby affecting expression of the coding genes. We showed that serum stimulation activates transcription of many cell growth–regulated genes in their coding region as well as the enhancer (e) sequences present either in upstream or downstream. Transcription of both coding and eRNAs were dependent on BRD4’s interaction with acetylated histones, given that JQ1, a BRD4 small molecule inhibitor, blocked both RNAs. In addition, ChIP-seq data showed that BRD4 localized to the coding and enhancer regions overlapping with the localization of Pol II, supporting the view that BRD4 takes part in the elongation processes by virtue of its ability to bind to acetylated histones. To confirm the broad role of BRD4 in RNA transcription, we labeled nascent RNA with Br-UTP and immunoprecipitated it with BRD4 antibody and then with BrdU antibody. Double immunoprecipitated RNA samples were analyzed by deep sequencing. We found that BRD4 is bound to newly synthesized eRNAs as well as to coding RNAs. We suggest a model wherein BRD4 directs elongation of eRNA and coding RNA (mRNA). The process is guided by the distribution of acetylated histones, highlighting an epigenetic component of transcription.
Analysis of Brd4 conditional knockout mice: BRD4 promotes proliferation by regulating many cell-cycle genes.
BRD4 is a bromodomain protein of the BET family that binds to acetylated histones. It is expressed at high levels in most, if not all cells. BRD4 is necessary for very early embryonic development. Thus conventional Brd4 knockout mice are embryonic lethal. We previously showed that BRD4 recruits P-TEFb and promotes transcription of cellular and viral genes. Recent reports found that BRD4 also has a critical role in forming super-enhancers. Super-enhancers are long stretches of regulatory DNAs densely occupied by transcription factors and chromatin regulators. They direct very strong transcription of select genes, thus helping define cell and lineage identity. The development of small molecule inhibitors that inhibit binding of acetyl-histones to the BET family proteins has had a dramatic effect on research on BRD4. The inhibitors, mostly by acting on BRD4 among other BET proteins, antagonize cancer growth, particularly leukemia and lymphoma, and inhibit inflammatory responses relevant to cardiovascular and autoimmune diseases. The findings implicate BRD4 for disease promotion but offer new therapeutic possibilities for a number of difficult-to-treat diseases. However, because of inherent limitations of pharmacological approaches, the precise role of BRD4 in healthy and diseased conditions remains uncertain. For example, the impact of BET inhibitors on normal cells is not well understood (such as on hematopoietic stem cells when treating leukemia/lymphoma), nor is it known how BET inhibitors affect anti-microbial activity of macrophages when used for treatment of inflammatory diseases. It is thus critical to gain full understanding of BRD4 activity in normal hematopoiesis and during innate and adaptive immunity.
Our approach is to investigate the role of BRD4 and its mechanism of action by studying Brd4 conditional knockout mice, rather than through the use of inhibitors. Currently, we are investigating the role of BRD4 in cell-cycle progression using mouse embryonic fibroblasts (MEFs) from Brd4f/fxERT2-Cre. In these cells, the Brd4 gene can be deleted in a Tamoxifen-inducible manner. Flow cytometry analysis of synchronized cells showed that cell-cycle progression is severely defective in Brd4–/– MEFs, in that G1/S transition and progression to G2/M are both delayed or blocked in a substantial fraction of cells (Figure 3, upper panel). The data indicate that BRD4 regulates cell growth at several stages by a complex, multi-faceted mechanism, beyond the regulation of c-Myc at G1, as proposed for cancer cells. Our RNA-seq data obtained from G0, G1, S, and G2/M stages substantiated this notion and showed that cell-cycle genes active at different stages are broadly downregulated in Brd4–/– cells. We surmise that BRD4 directly binds to many of these genes and drives their transcription independently. The view is supported by ChIP-seq analysis of genome-wide BRD4 binding at different stages of cell cycle, as BRD4 was found to occupy cell-cycle genes in a time-dependent manner. An IGV (Integrative Genomics Viewer) example in shows that BRD4 is absent from the histone H1 loci in G0, but occupies the loci 16 h after release (S phase) in wild-type cells, but not in Brd4–/– cells (KO) (Figure 3, lower panel). To eliminate false (phantom) peaks, ChIP-seq of Brd4–/– cells authenticates the BRD4 peaks. To further study BRD4 activity at multiple cell-cycle stages, particularly at G2/M, we plan to focus on several G2/M genes to delineate the role of BRD4 in maintaining genome integrity.
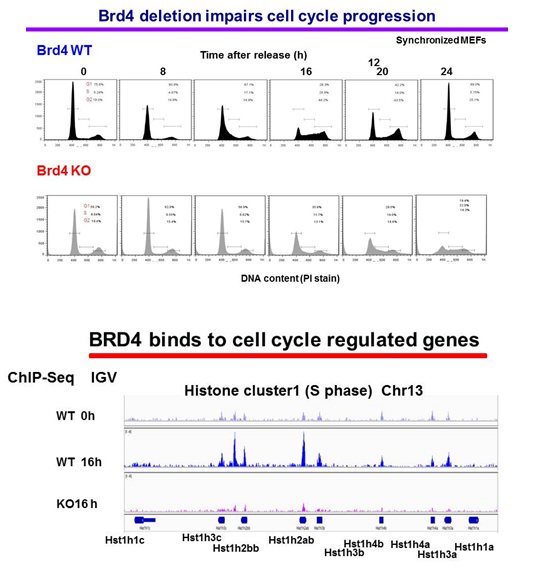
Click image to enlarge.
Figure 3. BRD4 is required for cell-cycle progression of normal cells.
Upper panel: wild-type (WT) and Brd4–/– (KO) mouse embryonic fibroblasts (MEFs) were synchronized by serum starvation, and cell-cycle progression was examined after release. Propidium iodide staining shows percentages of cells in G1. S, G2/M.
Lower panel: BRD4 binding was tested by ChIP-seq analysis of synchronized WT and Brd4 KO cells. The figure depicts BRD4 occupancy at the histone H1 locus.
Publications
- Ayithan N, Bradfute S B, Anthony SM, Stuthman KS, Bavari S, Bray M, and Ozato, K. Virus-like particles activate type I interferon pathways to facilitate post-exposure protection against Ebola virus infection. PLoS One 2015;10:e0118345.
- Gupta M, Shin DM, Ramakrishna L, Goussetis DJ, Platanias LC, Xiong H, Morse HC 3rd, Ozato K. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat Commun 2015;6:6379.
- Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, Dey A, Ozato K, Singer DS. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol 2016;23:540-548.
- Chang TH, Yoshimi R, Ozato K. Tripartite Motif (TRIM) 12c, a mouse homolog of TRIM5, is a ubiquitin ligase that stimulates Type I IFN and NF-kB pathways along with TNFR-associated Factor 6. J Immunol 2015;195:5367-5379.
- Bachu M, Dey A, Ozato K. Chromatin landscape of the IRF genes and role of the epigenetic reader BRD4. J Interferon Cytokine Res 2016;36:470-475.
Collaborators
- Steven Holland, MD, Laboratory of Clinical Infectious Diseases, NIAID, Bethesda, MD
- Herbert Morse II, MD, Laboratory of Immunopathology, NIAID, Rockville, MD
- Dinah S. Singer, PhD, Experimental Immunology Branch, NCI, Bethesda, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
- Jun Zhu, PhD, DNA Sequencing and Genomics Core, NHLBI, Bethesda, MD
Contact
For more information, email ozatok@mail.nih.gov or visit http://ozatolab.nichd.nih.gov.