Receptors and Actions of Peptide Hormones and Regulatory Proteins in Endocrine Mechanisms

- Maria L. Dufau, MD, PhD, Head, Section on Molecular Endocrinology
- Raghuveer Kavarthapu, PhD, Staff Fellow
- Peng Zhao, PhD, Postdoctoral Fellow
We investigate the molecular basis of peptide hormone control of gonadal function, with particular emphasis on the structure and regulation of the genes encoding the luteinizing hormone receptor (LHR) and prolactin (PRL) receptor (PRLR). We also investigate the regulatory mechanism(s) involved in the progression of spermatogenesis and the control of Leydig cell (LC) function. Our studies focus on the regulation of human LHR transcription (nuclear orphan receptors, epigenetics, DNA methylation, second messengers, repressors, corepressors, and coactivators), as well as on the multiple-promoter control of hPRLR gene transcription. We are elucidating the relevance of PRL, estradiol and its receptor (liganded or un-liganded), epidermal growth factor (EGF), the EGF receptors ERRBB1/EGFR and ERRB2/HER2 in the up-regulation of the PRLR, and their mechanistic commonalities for definition of PRL/PRLR–induced progression and metastasis of breast tumors and their role in persistent invasiveness in certain states refractory to adjuvant endocrine therapies. We also investigate novel gonadotropin-regulated genes relevant to the progression of testicular gametogenesis, LC function, and other endocrine processes. We focus on the function and regulation of the gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), an essential post-transcriptional regulator of spermatogenesis, which was discovered, cloned, and characterized in our laboratory. The various functions of GRTH/DDX25 provide a fertile ground for the development of a male contraceptive.
The luteinizing hormone receptor
LHR is expressed primarily in the gonads, where it mediates LH signals, which regulate either cyclic ovarian changes or testicular function. The LHR transcription gene is controlled by complex and diverse networks, in which coordination and interactions between regulatory effectors are essential for silencing/activation of LHR expression. The proximal promoter site for the transcription factor Sp1 recruits histone (H) deacetylases (HDAC) and the Sin3A (a transcriptional regulatory protein) corepressor complex, which contributes to the silencing of LHR transcription. Site-specific acetylation/methylation-induced phosphatase release serves as an on switch for Sp1 phosphorylation at Ser641, which causes p107 repressor release from Sp1, recruitment of Transcription Factor II B (TFIIB) and RNA polymerase II (Pol II), and transcriptional activation. Maximal derepression of the gene is dependent on DNA demethylation of the promoter, on H3/acetylation, and on HDAC/Sin3A release. Positive Cofactor 4 (PC4) has an important role in the formation assembly of the preinitiation complex (PIC) in trichostatin A (TSA)–mediated LHR transcription. It is recruited by Sp1 following TSA treatment and acts as a coactivator. However, PC4 does not participate in TSA release of phosphatases, Sp1 phosphorylation, or release of repressor or complexes. Although TFIIB recruitment is dependent on PC4, we ruled out TFIIB, as its direct target, and acetylation of PC4 in the activation process. However, we demonstrated TSA–induced acetylation of PC4–interacting proteins, identified as acetylated H3 by mass spectrometry, and PC4's presence in the complex, in association with chromatin at the promoter, was demonstrated by ChIP/reChIP. The role of these interactions on chromatin structure and their participation in the assembly of the PIC and transcriptional activation are under investigation. To elucidate the physiological impact of PC4 on Sp1–directed transcription in gonads, we generated a cell-specific knockout (KO) by breeding PC4–floxed mice with transgenic mice expressing tissue-specific Cyp17 Cre. We are now analyzing PC4 null-mice with a specific deletion in testicular Leydig cells.
Gonadotropin-regulated testicular RNA helicase
Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) is a testis-specific member of the DEAD-box family of RNA helicases present in Leydig cells (LC) and meiotic germ cells and is essential for the completion of spermatogenesis. Males lacking GRTH are sterile owing to the absence of sperm resulting from failure of round spermatids to elongate. Besides its intrinsic RNA helicase activity, it is a shuttling protein that exports specific mRNAs from the nucleus to cytoplasmic sites. Our studies demonstrated the essential participation of GRTH–mediated export/transport of mRNAs in the structural integrity of the Chromatoid Body (storage/processing of mRNAs) and of their transit/association to actively translating polyribosomes, where GRTH may regulate translational initiation of genes. GRTH is regulated by LH through androgen at the transcriptional level in LCs (directly), with impacts in steroidogenesis, and germ cells (indirectly via androgen receptor in Sertoli cells); GRTH's expression is both cell- and stage-specific. The helicase displays negative autocrine control of androgen production in LCs by preventing overstimulation of the LH–induced androgen pathway through enhanced degradation of the StAR protein (steroidogenic acute regulatory protein). In this manner, GRTH controls the degree of cholesterol transport to the mitochondria and its availability for steroidogenesis. Transgenic mouse models carrying a GRTH 5′ flanking region–GFP reporter provided in vivo systems that permit differential elucidation of regions in the GRTH gene that direct the gene's expression (upstream; –6.4/–3.6kb) in germ cells (pachytene spermatocytes and round spermatids) and downstream in LCs as well as of the gene's direct regulation by androgen/androgen receptor (A/AR) in LCs and indirect regulation in germ cells.
A functional binding site for the germ cell–specific transcription factor Germ Cell Nuclear Factor (GCNF) resides in the distal region, –5270/–5252, of the GRTH gene, and we identified its paracrine regulation by androgen at this site (Reference 3), which is cell-specific and occurs exclusively in round spermatids (RS). GCNF is a member of the orphan nuclear receptor superfamily that is expressed in the nucleus of spermatocytes and spermatids of adult mice. GCNF is essential for GRTH expression in germ cells. GCNF knock-down in round spermatids preparations from testis of transgenic mice reduced GFP/GRTH expression upon in vivo treatment of mice with Flutamide (Flu), an AR antagonist. Moreover, Flu treatment of wild-type mice caused selective reduction of GRTH in round spermatids. The studies provided evidence for actions of androgen on GCNF cell-specific regulation of GRTH expression in germ cells. Also, GRTH associates with GCNF mRNA, and its absence causes an increase in GCNF expression and mRNA stability, indicative of negative autocrine regulation of GCNF by GRTH. Our in vivo and in vitro models link paracrine androgen action with two relevant germ-cell genes essential for the progress of spermatogenesis and established their regulatory interrelationship. The studies provide valuable insights and facilitate what could be a difficult search for androgen/androgen receptor–mediated gene products in Sertoli cells that affect germ cell function and spermatogenesis.
The prolactin receptor
The human prolactin receptor (PRLR) mediates the diverse cellular actions of prolactin (PRL). PRL plays a major role in the proliferation and differentiation of breast epithelium and is essential for stimulation and maintenance of lactation. It plays an important role in the etiology and progression of breast cancer, tumoral growth, and chemo-resistance. PRLR expression is controlled at the transcriptional level by several promoters, one generic (PIII), which lacks an estrogen response element (ERE) and is preferentially utilized, and five human-specific (hPN1–hPN5), which we defined and characterized in our laboratory. Each promoter directs transcription/expression of a specific non-coding exon 1, a common non-coding exon 2, and coding exons (E3–E11). Complex formation of the estrogen receptor alpha (ERa) homodimer (non-DNA bound) with the transcription factor Sp1 and dimers of the transcription factor C/EBPβ, bound to their sites at the PIII promoter, is required for basal (constitutive ERa homodimers) and estradiol (E2)–induced transcriptional activation/expression of the human PRLR gene.
In tumoral breast, PRL causes cell proliferation by activating its cognate receptor. Exacerbation of PRL's actions in breast cancer, resulting from increased receptor expression, can explain resistance to estrogen inhibitors in breast cancer. Using MCF7 breast cancer estrogen-responsive (ER+)/oncogene HER2–positive (HER2+) cells, we recently demonstrated that, in the absence of E2, exogenous/endogenous PRL upregulated PRLR transcription/expression, with essential participation of ERa and of the JAK2/STAT5, mitogen-activated protein kinase (MAPK), and PI3K pathways. This occurs by interaction of phosphorylated ERa (generated by PRL/PRLR/JAK2) associated with Sp1 and C/EBPβ with STAT5A and STAT5B bound to a nucleotide sequence, known as a GAS element, in the PIII promoter. We also found that ERRB/HER2, which is expressed in 10% of breast tumors, phosphorylated and activated by JAK2 via PRL/PRLR, induces ERa phosphorylation. Such cross-talk activation of ERBB2/HER2 signaling was identified as an alternate route for the PRLR increase, which is abolished by mutation of the GAS site (Stat5 DNA-recognition motif), by Stat5 siRNA, or by an ER antagonist (ICI). This indicates that ERa participates in PRLR transcription via PRL/PRLR/Stat5. PRL/PRLR induces phosphorylation of ERa through the JAK2/PI3K/MAPK/ERK– and HER2–activated pathways (Reference 1). Increased recruitment of phospho-ERa to Sp1 and C/EBPβ bound at promoter sites is essential for PRL–induced receptor transcription. Direct evidence for local actions of PRL independent of E2 is provided by the up-regulation of PRLR transcription/expression via the Stat5/ERa activation loop, with requisite participation of signaling mechanisms. These studies, which demonstrated a central role ERa in PRLR receptor up-regulation, are of relevance in states refractory to aromatase inhibitors, in which cancer progression can be fueled by endogenous PRL. Therapies that inhibit the function of PRL or PRLR, combined with inhibitors of various signaling pathways, could reverse resistance in breast cancer. Moreover, a combination therapy targeting ER and PRLR directly can offer an additional avenue to eliminate constitutive activation of ER and of PRLR by endogenous prolactin.
Paracrine inputs have an active role in breast tumor development, progression, and metastasis. Stromal fibroblasts secrete epidermal growth factor (EGF), which, through its receptor EGFR/ERRB1 present in breast tumors cells, activates signaling pathways, which in turn trigger requisite transcription factors and coactivators that can affect the proliferation of breast tumor cells. Our recent work addressed the role of EGFR in the up-regulation of the PRLR, given that most breast cancers that become resistant to endocrine therapy have elevated expression/activation of EGFR and its family member ERBB2. We showed, in MCF7 cells, marked activation of PRLR gene transcription/expression by exogenous EGF independent of PRL/PRLR/JAK2 or E2, with essential involvement of the MAPK, ER1/2, and PI3K-AKT signaling pathways (Reference 5). For their recruitment to the PRLR PIII promoter, these are mediated by EGFR tyrosines 1068 and 1086 and the tyrosine kinase cSRC–dependent EGFR tyrosine 845 for ERa and STAT5b phosphorylation, respectively (Figure 1). Apart from its independence of E2 and the activator requirements (PRL vs. EGF), there are important commonalities (prerequisite for ERa and STAT5) in the mechanism of PRLR transcription/expression. Moreover, the studies revealed that STAT 5 interaction with ERa is essential for PRLR up-regulation. Our findings provide mechanistic avenues whereby, upon resistance to hormonal therapy, an increase of PRLR could promote progression and metastasis in breast cancer.
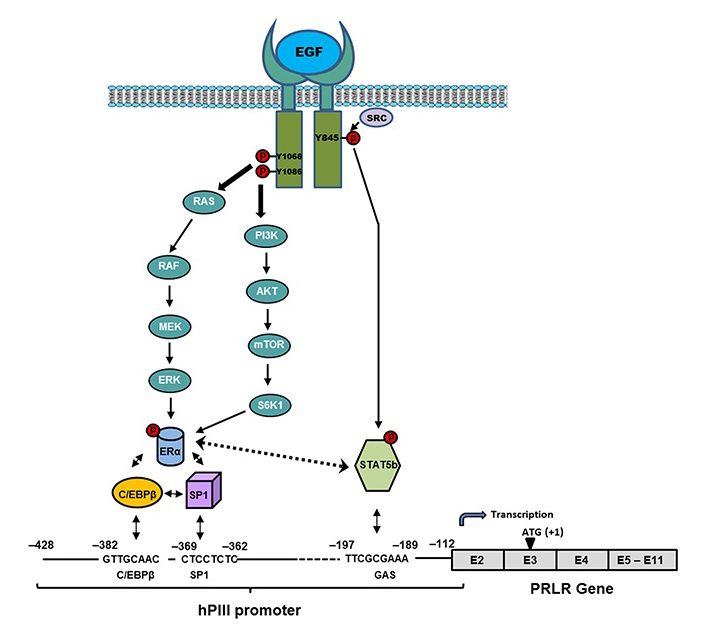
Click image to enlarge.
Figure 1. Mechanism of up-regulation of the prolactin receptor (PRLR) by epidermal growth factor (EGF)/epidermal growth factor receptor ERRB1 (EGFR) independent of estradiol and endogenous prolactin
Signal transduction mechanisms induced by EGF via EGFR, causing activation of transcription factors that are required for PRLR gene activation through the hPIII (human PIII) promoter of PRLR and consequent increased expression of the receptor in MCF-7 cells
Publications
- Kavarthapu R, Tsai Morris CH, Dufau ML. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERa and STAT5. Oncotarget 2014;5:9079-9091.
- Yang, R, Tsai-Morris CH, Kang JH, Dufau ML. Elucidation of RNA binding regions of gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) to transcripts of a chromatin remodeling protein essential for spermatogenesis. Horm Mol Biol Clin Investig 2015;22:119-130.
- Kavarthapu R, Dufau ML. Germ Cell Nuclear (GCNF/RTR) regulates transcription of gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) in testicular germ cells—the androgen connection. Mol Endocrinol 2015;29:1792-1804.
- Chason RJ, Kang J-H, Gerkowicz SA, Dufau ML, Catt KJ, Segars JH. GnRH agonist reduces estrogen receptor dimerization in GT1-7 cells: Evidence for cross-talk between membrane-initiated estrogen and GnRH signaling. Mol Cell Endocrinol 2015;404:67-74.
- Kavarthapu R, Dufau ML. Role of EGF/ERBB1 in the transcriptional regulation of the prolactin receptor independent of estrogen and prolactin in breast cancer cells. Oncotarget 2016;7:65602-65613.
Collaborators
- James M. Pickel, PhD, Transgenic Core Facility, NIMH, Bethesda, MD
- James H. Segars, MD, Unit on Reproductive Endocrinology and Infertility, NICHD, Bethesda, MD
Contact
For more information, email dufau@helix.nih.gov or visit irp.nih.gov/pi/maria-dufau.