Molecular Nature and Functional Role of Dendritic Voltage-Gated Ion Channels

- Dax Hoffman, PhD, Head, Molecular Neurophysiology and Biophysics Section
- Jiahua Hu, PhD, Staff Scientist
- Lin Lin, PhD, Microbiologist
- Ying Liu, MD, Biologist
- Jakob Gutzmann, PhD, Visiting Fellow
- Erin Gray, PhD, Postdoctoral Fellow
- Jon Murphy, PhD, Postdoctoral Research Associate Program (PRAT) Fellow
- Travis Tabor, BS, Postbaccalaureate Fellow
The central nervous system underlies all our experiences, actions, emotions, knowledge, and memories. With billions of neurons each firing hundreds of times per second, the complexity of the brain is stunning. To pare down the task of understanding something so complex, our research approach calls for studying the workings of a single central neuron—the pyramidal neuron from the CA1 region of the hippocampus. The hippocampus is essential for long-term memory in humans and is among the first brain regions affected by epilepsy and Alzheimer’s disease. To understand how the hippocampus stores and processes information, we focus on one of its principal cell types, the CA1 pyramidal neuron. Each pyramidal neuron in the CA1 region of the hippocampus receives tens of thousands of inputs onto its dendrites, and it is commonly thought that information is stored by altering the strength of individual synapses (synaptic plasticity). Recent evidence suggests that the regulation of synaptic surface expression of glutamate receptors can, in part, determine synaptic strength. However, the dendrites contain an abundance of ion channels that are involved in receiving, transforming, and relaying information in the dendrites, adding an additional layer of complexity to neuronal information processing.
We found that the A-type potassium channel subunit Kv4.2 is highly expressed in the dendritic regions of CA1 neurons in the hippocampus and, as one of the primary regulators of dendritic excitability, plays a pivotal role in information processing. Kv4.2 is targeted for modulation during the types of plasticity thought to underlie learning and memory. Moreover, we found that the functional expression level of Kv4.2 regulates the subtype expression of NMDA–type glutamate receptors, the predominant molecular devices controlling synaptic plasticity and memory. We are currently following up on these findings with more detailed investigations into the mechanisms of activity-dependent Kv4.2 regulation. In addition, we have begun investigating the role of dendritic voltage-gated channels in neuronal development and developmental disorders.
Role of voltage-gated ion channels in synaptic development and disease
Calcium regulation of transient K+ currents
The sub-threshold activating, A-type potassium current (IA) has long been recognized as a critical component for regulating dendritic excitability and shaping back-propagating action potentials. In CA1 pyramidal cells of the hippocampus IA is carried predominantly by Kv4.2 potassium channels, which are associated with a variety of accessory proteins that modulate their trafficking and kinetics. Among these interacting proteins are K-channel–interacting proteins (KChIPs), small cytoplasmic proteins with four calcium-binding EF-Hand motifs and dipeptidyl-peptidase–like proteins (DPPs). IA shows a distinct functional gradient along the apical dendrites of CA1 pyramidal neurons. The reciprocal relationship between Kv4.2 and the GluN2B subunit of the N-methyl-D-aspartate (NMDA) receptor, and its interaction with the voltage-gated calcium channel Cav2.3 opens up the possibility of calcium-dependent effects on Kv4.2 function in the synapse. Using a mutated KChIP construct that is unable to bind to calcium but still interacts with Kv4.2, Jakob Gutzmann found that changes in intracellular calcium concentration alters many Kv4.2–mediated current properties in the presence of KChIPs in a heterologous expression system. All these effects were abolished when we used a KChIP2a construct whose ability to bind to calcium was mutated. Performing these experiments in the more physiological environment of acute hippocampal slices led to similar results. We are now recording IA from somatic and dendritic outside-out patches in mice, in order to better understand the interplay between calcium and potassium that shapes CA1 pyramidal cells' electrical behavior.
Isomerase regulation of potassium channel trafficking and function
To identify Kv4.2 binding proteins, Jiahua Hu employed a tandem affinity purification approach (TAP) to isolate the Kv4.2 protein complex from hippocampal neurons. Mass-spectrometry analysis identified known proteins such as KChIP family members and DPP6/10. The TAP–MS assay also identified an isomerase as a binding partner of Kv4.2. The binding was confirmed by brain co-immunoprecipitation, co-expression in HEK293T cells, and peptide pull down in vitro. The isomerase binds to a specific Kv4.2 site, and the association is regulated by neuronal activity and seizure. The isomerizing activity may regulate Kv4.2 binding to its auxiliary subunits, suggesting that the isomerase plays a role in Kv4.2 function. To further study the physiological function of isomerase and Kv4.2 channel, we are generating a knockin mouse in which the isomerase binding site is specifically abolished.
DPP6 deletion leads to memory impairments in mice.
We previously showed that the Kv4 A-type K+ auxiliary subunit DPP6 plays important roles hippocampal synaptic development and function but that the behavioral effects of DPP6–KO (knockout) mice are unknown. Lin Lin focused on hippocampus-dependent tasks involved in recognition and learning memory. We found DPP6–KO mice to be impaired in recognition memory with a Novel Object Recognition task. In the Water Maze, DPP6–KO mice showed slow learning and impaired memory performance. In a T-Maze task, the success rate of wild-type (WT) mice rose over the course of the training, while DPP6–KO mice started and finished with the same success rate without any significant learning throughout the training. Taken together, we find that DPP6 deletion leads to behavioral impairments in recognition and spatial learning and memory. We are investigating whether the behavioral deficits can be changed or rescued by DPP6 microinjected by in utero electroporation (IUE) in DPP6–KO mice.
Loss of regulated Cav2.3 expression in a mouse model of Fragile X syndrome
Fragile X syndrome (FXS) is a severe form of intellectual disability in humans, which arises from the loss of the fragile X mental retardation protein (FMRP), an mRNA–binding protein that regulates translation downstream of group I metabotropic glutamate receptors (GpI mGluRs). Loss of FMRP leads to enhanced calcium spiking and neuronal excitability. Erin Gray therefore sought to explore the possibility that FMRP regulates expression of the dendritic voltage-gated calcium channel Cav2.3. We had found previously that the cortices of FMRP–KO mice exhibit enhanced levels of Cav2.3 under basal conditions, and we were curious to know how loss of FMRP might affect Cav2.3 expression in the hippocampus. We performed Western blotting to measure Cav2.3 protein levels from either acutely isolated or cultured hippocampal neurons and, similar to our previous findings in cortex, we found that the FMRP–KO mice exhibit enhanced expression of Cav2.3 in the hippocampus. The increase in Cav2.3 expression should impact neuronal physiology; therefore, we isolated Cav2.3 currents using whole-cell electrophysiology in cultured hippocampal neurons from WT and FMRP–KO mice. As predicted from our Western blot data, FMRP–KO neurons exhibited greatly enhanced Cav2.3 current amplitudes. Thus, it appears that FMRP normally represses Cav2.3 translation under basal conditions and that loss of FMRP leads to an increase in Cav2.3 protein in the membrane. We are also investigating the possibility that the FMRP repression of Cav2.3 expression can be regulated by upstream activity of GpI mGluRs. In support of this idea, our previous data showed that stimulation of GpI mGluRs does indeed increase Cav2.3 translation and expression in WT neurons but not in neurons lacking FMRP. Next, to determine whether FMRP regulates translation of existing Cav2.3 mRNA or whether transcription is required, we applied the transcriptional blocker Actinomycin D (ActD) to cultured neurons while stimulating GpI mGluRs. Treating neurons with ActD did not significantly inhibit the stimulation of Cav2.3 expression, suggesting that GpI mGluRs raise Cav2.3 protein by inducing translation of existing mRNA bound to FMRP. Taken together, our data suggest that FMRP represses translation of Cav2.3 under basal conditions and that activation of GpI mGluRs disinhibits FMRP to allow for active translation of Cav2.3. The loss of regulated Cav2.3 expression seen in the FMRP KO may underlie the atypical calcium signaling and neuronal excitability seen in FXS and may thus be a potential therapeutic target in this disease.
FMRP interacts with Cav2.3 mRNA.
Ying Liu showed that Cav2.3 mRNA levels were altered in FMRP–KO neurons and that Cav2.3 protein levels were significantly enhanced in the FMRP KO. To determine whether Cav2.3 mRNA is one of the targets of FMRP, we performed RNA immunoprecipitation. Our data showed that FMRP interacts with Cav2.3 mRNA in transfected HEK293 cells and in mouse cortex and hippocampus. Our results suggest that FMRP binds to Cav2.3 mRNA directly or indirectly to repress Cav2.3 translation and regulate neuronal excitability.
In related work, using the FMRP–KO mouse, Jon Murphy is examining whether dendritic FMRP regulates mRNA trafficking and protein expression of CaV2.3 and Kv4.2 in the dendrites of hippocampal neurons (Figure 1). Recent work centered primarily on analysis of mRNA localization and regulation of total protein translation in neuronal dendrites of WT and FMRP–KO mouse neurons. Using fluorescence in situ hybridization to detect mRNAs for CaV2.3 and CaMKII (Ca2+/calmodulin-dependent protein kinase II) in neurons, we have found that the abundance of CaMKII mRNA, which encodes a known dendritically synthesized protein, is elevated throughout the dendritic arbor of FMRP–KO mice. FMRP is known to inhibit CaMKII translation through direct binding to CaMKII mRNA, suggesting that either CaMKII mRNA is more highly transcribed in FMRP–KO mice, or that it is no longer sequestered in mRNA–protein complexes where in situ hybridization is inhibited, or both. Conversely, CaV2.3 mRNA signals are low throughout dendrites in both WT and FMRP–KO neurons. Studies of Kv4.2 mRNA localization in WT and FMRP–KO neurons are ongoing.
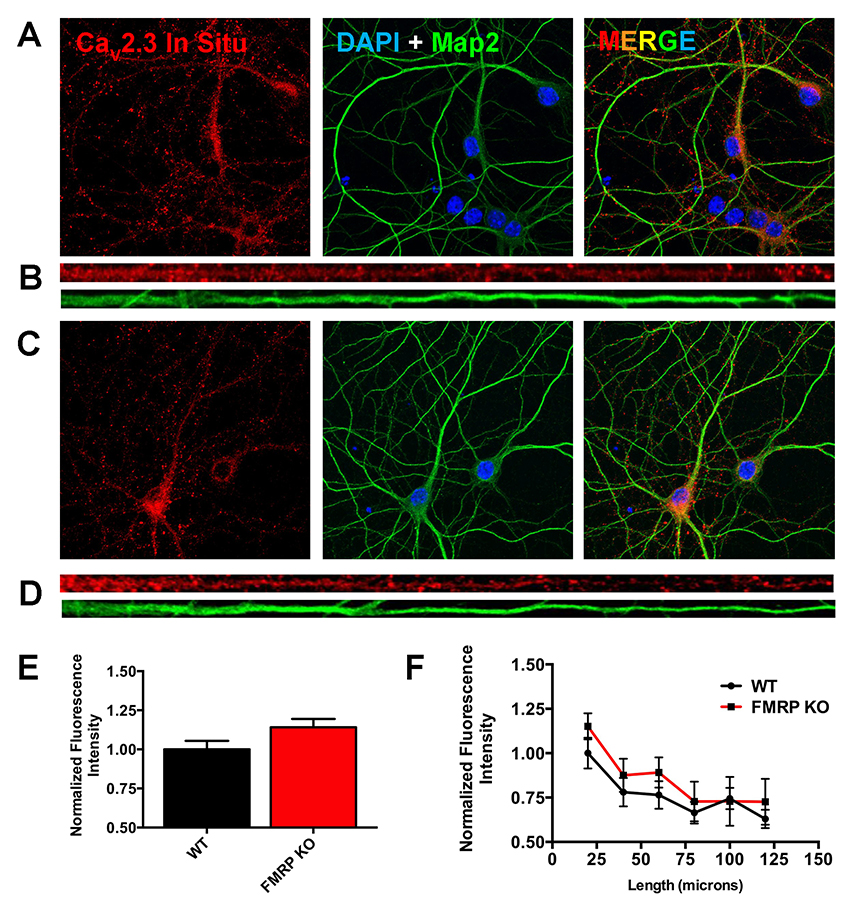
Click image to enlarge.
Figure 1. Elevated levels of CaV2.3 mRNA can be detected in the cell body and proximal dendrites in a mouse model of Fragile X syndrome.
A) Maximum intensity projection photomicrographs of WT mouse neurons showing Cav2.3 mRNA fluorescence in situ hybridization signal (left, red), overlaid nucleus (DAPI), and dendrite (Map2) cyto-architectural markers (middle), and a three-color composite image (right). B) Straightened dendrites demonstrating CaV2.3 mRNA transcript in situ staining (red) in a dendrite identified by Map2 immuno-cytochemistry (green). C) Maximum intensity projection photo-micrographs of FMRP–KO mouse neurons arranged as in “A”. D) Straightened dendrites of FMRP–KO mouse neurons arranged as in “B”. E) Comparison of normalized mean fluorescence intensity of CaV2.3 mRNA in situ measured at the neuronal cell body of WT and FMRP–KO mouse neurons. F) Comparison of the mean fluorescence intensity of CaV2.3 mRNA in situ in 20–micron bins extending from the neuronal cell body.
Cellular and molecular phenotypes associated with idiopathic autism in iPSC-derived neurons
Idiopathic autism is an early onset neurodevelopmental disorder that may result from genetic variation in multiple genes. We generated induced pluripotent stem cells (iPSCs) from three patients with intrinsic autism as well as from controls (Figure 2); the autistic iPSCs were subsequently differentiated into electrophysiologically active neurons comparable to control cells. Autistic iPSC–derived neurons displayed significant decreases in both Na+ and fast K+ voltage-gated currents but not in Ca2+ and slow K+ voltage-gated currents. The defects accounted for the observation that autistic iPSC–derived neurons reach faster action potential saturation and are slightly more excitable than controls. We found that the amplitude and frequency of spontaneous excitatory postsynaptic currents from autistic neurons were significantly lower than in controls, indicating defects in excitatory synaptic transmission (Reference 4).
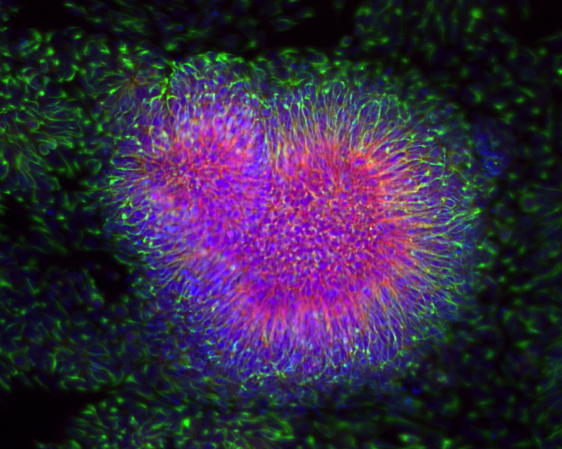
Click image to enlarge.
Figure 2. Cellular and molecular phenotypes associated with idiopathic autism in iPSC–derived neurons
Immuno-fluorescence images of human induced pluripotent stem cell (iPSCs)–derived neuronal progenitor cells showing “neural rosette structure,” where neuro-epithelial cells arrange radially around a lumen. Cells were stained with PAX6 (red), NESTIN (green) and DAPI (blue).
Additional Funding
- Postdoctoral Research Associate Program (PRAT) Fellowship (Jon Murphy)
Publications
- Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, Hoffman DA, Roberson ED. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease. J Neurosci 2015;35(15):6221-6230.
- Murase S, Lantz CL, Kim E, Gupta N, Higgins R, Stopfer M, Hoffman DA, Quinlan EM. Matrix metalloproteinase-9 regulates neuronal circuit development and excitability. Mol Neurobiol 2015;53(5):3477-3493.
- Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015;7(307):307ra153.
- Liu X, Campanac E, Cheung HH, Ziats MN, Canterel-Thouennon L, Raygada M, Baxendale V, Pang AL, Yang L, Swedo S, Thurm A, Lee TL, Fung KP, Chan WY, Hoffman DA, Rennert OM. Idiopathic autism: cellular and molecular phenotypes in pluripotent stem cell-derived neurons. Mol Neurobiol 2016;Epub ahead of print.
- Sertedaki A, Markou A, Vlachakis D, Kossida S, Campanac E, Hoffman DA, Sierra ML, Xekouki P, Stratakis CA, Kaltsas G, Piaditis GP, Chrousos GP, Charmandari E. Functional characterization of two novel germline mutations of the KCNJ5 gene in hypertensive patients without primary aldosteronism but with ACTH-dependent aldosterone hypersecretion. Clin Endocrinol (Oxf) 2016;85(6):845-851.
Collaborators
- Sachiko Murase, PhD, Laboratory of Molecular Biology, NINDS, Bethesda, MD
- Avindra Nath, MD, Translational Neuroscience Center, NINDS, Bethesda, MD
- Owen M. Rennert, MD, Laboratory of Clinical Genomics, NICHD, Bethesda, MD
- Eric Roberson, MD, PhD, University of Alabama, Birmingham, AL
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Paul Worley, MD, The Johns Hopkins University, Baltimore, MD
Contact
For more information, email hoffmand@mail.nih.gov or visit http://hoffmanlab.nichd.nih.gov.