Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Ingo Janssen, MD, Postdoctoral Visiting Fellow
- Petra Bullova, MS, Predoctoral Visiting Fellow
- Ivana Jochmanova, MD, Predoctoral Visiting Fellow
- Roland Darr, MD, Volunteer
- Garima Gupta, MD, Volunteer
- Veronika Caisova, MS, Predoctoral Visiting Fellow
- Katherine Wolf, BS, Postbaccalaureate Fellow
- Bruna Viana, BS, Postbaccalaureate Fellow
- Ying Pang, MD, PHD, Postdoctoral Visiting Fellow
- Jean Caringal, MD, Volunteer
- Amy Lopez, MD, Volunteer
- Valerie Valdez, MD, Volunteer
- Erick Mendoza, MD, Volunteer
- Julie Zemskova, MS, Summer Student
- Gabriel Schneider, Summer Student
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma (PHEO) and paraganglioma (PGL). Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and 'reverse translation research,' by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to (1) establish new and improved methods and strategies for novel diagnostic and localization approaches to PHEO/PGL; (2) explain the molecular and cellular basis for varying clinical presentations of PHEOs/PGLs and establish the pathways of tumorigenesis; (3) search for new molecular and genetic/epigenetic markers for diagnosis and treatment of metastatic PHEO/PGL; (4) introduce new therapeutic options for malignant/metastatic PHEO/PGL; and (5) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we enter into multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis/genetics and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
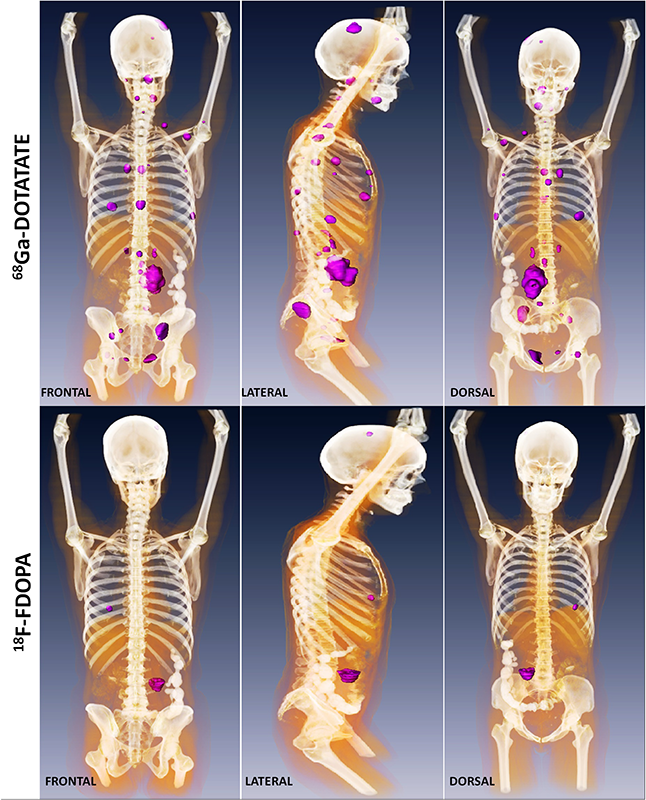
Click image to enlarge.
Metastatic paraganglioma detected by 68Ga-DOTATATE PET/CT and 18F-FDOPA PET/CT
Detection of metastatic paraganglioma with the novel imaging modality 68Ga-DOTATATE PET/CT compared with 18F-FDOPA PET/CT (frontal, lateral, and dorsal views)
Authors: Papadakis GZ, Bagci U, Millo CM, Janssen I, Patronas NJ, Stratakis CA, Pacak K
Clinical aspects of pheochromocytoma and paraganglioma
Overall, about 10 to 20% of PHEOs/PGLs (also collectively known as PPGLs), are metastatic, with higher metastatic potential observed in succinate dehydrogenase subunit B/fumarate hydratase (SDHB/FH)–related tumors. Owing to the improved availability of biochemical and genetic testing and the frequent use of anatomical/functional imaging, there is currently an improved detection rate of metastatic PPGL. We conducted a retrospective analysis of 132 patients (27 children, 105 adults) with metastatic PPGL, diagnosed and treated from 2000 to 2014. Seventy-seven (58%) males and 55 (42%) females were included; 39 (30%) have died, with no sex preference. Seventy-three (55%) patients had SDHB mutations; 59 (45%) patients had apparently sporadic tumors (AST). The average age of SDHB patients at primary tumor diagnosis (31 ± 16 years) was significantly lower than AST patients (40 ± 15 years). The average metastatic interval (MI) declined significantly with increasing age in both SDHB and AST patients. Only 16% of all primary tumors were smaller than 4.5 cm. Eleven percent of patients had biochemically silent disease, more with SDHB. Of SDHB patients, 23% had metastatic tumors at first diagnosis, compared with 15% of AST patients. Five- and 10-year survival rates were significantly better for metastatic AST than for SDHB patients. Overall, survival was significantly longer in children than in adults, especially in SDHB patients. The deceased patients all died from PPGL, whose phenotypes were mainly noradrenergic. We concluded, that, in children, metastatic PPGLs are mainly the result of SDHB mutations; in adults they are equally distributed between in SDHB mutations and AST, with better 5- and 10-year survival rates for ASTs. Among SDHB patients, children survive longer than adults. Primary metastatic tumors, mostly presenting as noradrenergic PGLs, are larger than 4.5 cm in more than 80% of patients. The frequency of metastatic tumors from primary AST increases with age, including a lower MI compared with SDHB tumors.
Pharmacological treatment is mandatory in patients with hormonally functional PPGL. We evaluated whether patients initially diagnosed with hormonally functional PPGL by various medical subspecialties received proper adrenoceptor blockade, and we analyzed factors predicting the prescription of adequate treatment. In a retrospective cohort study, we reviewed data of 381 patients from Cedars-Sinai Medical Center and others outside the National Institutes of Health initially diagnosed with hormonally functional PPGL, who were referred to these institutions between January 2001 and April 2015. We used logistic regression to assess factors associated with proper adrenoceptor blockade. Adequate pharmacological treatment was prescribed to 69·3%, of which 93·1% received α-adrenoceptor blockers. Regarding patients who were inappropriately treated, 53% did not receive any medication. Independent predictors of the prescription of a proper blockade were the diagnosis by endocrinologists, the presence of high blood pressure, and evidence of metastasis. We concluded that, although the majority of patients received adequate pharmacological treatment, almost one-third were either not treated or received inappropriate medications. The diagnosis by endocrinologists, the presence of high blood pressure, and the evidence of metastatic disease were identified as independent predictors of a proper blockade. The results highlight the need to educate physicians about the importance of starting adequate adrenoceptor blockade in all patients with hormonally functional PPGL.
Hereditary pheochromocytoma and paraganglioma
The syndrome of PGL, somatostatinoma (SOM), and early childhood polycythemia in patients with somatic mutations in the hypoxia-inducible factor 2 alpha (HIF2A) gene has been described in only a few patients worldwide. The study provided detailed information about the clinical aspects and course of seven patients with this syndrome and brought these experiences into perspective with the pertinent literature. Six females and one male presented at a median age of 28 years (range 11–46). Two were found to have HIF2A somatic mosaicism. No relatives were affected. All patients were diagnosed with secondary polycythemia before age 8 and before PGL/SOM developed. PGLs were found at a median age of 17 years (range 8–38) and SOMs at 29 years (range 22–38). PGLs were multiple, recurrent, and metastatic in 100%, 100%, and 29% of all cases, and SOMs in 40%, 40%, and 60%, respectively. All PGLs were primarily norepinephrine-producing. All patients had abnormal ophthalmologic findings and those with SOMs had gallbladder disease. Computed tomography (CT) and magnetic resonance imaging (MRI) revealed cystic lesions at several sites and hemangiomas in four patients (57%), previously thought to be pathognomonic for von Hippel-Lindau disease. The most accurate radiopharmaceutical to detect hereditary PGL associated with polycythemia appeared to be [18F]-fluorodihydroxyphenylalanine ([18F]-FDOPA). Therefore, [18F]-FDOPA PET/CT, but not [68Ga]-(DOTA)-[Tyr3]-octreotate ([68Ga]-DOTATATE) PET/CT, is recommended for tumor localization and aftercare in this syndrome. The long-term prognosis of the syndrome is unknown. However, to date no deaths occurred after six years follow-up. Physicians should be aware of this unique syndrome and its diagnostic and therapeutic challenges.
Previously, mutations in the SDHA/SDHB/SDHC/SDHD, SDHAF2, VHL (encoding the von Hippel-Lindau tumor suppressor), FH, PHD1, and PHD2 genes (encoding hypoxia-inducible factor prolyl hydroxylases 1 and 2, respectively) have been associated with HIF (hypoxia-inducible factor) activation and the development of pseudo-hypoxic (cluster-1) PGLs. To a certain extent, these tumors overlap in terms of tumor location, syndromic presentation, and noradrenergic phenotype. However, they also differ particularly by clinical outcome and by the presence of other tumors or abnormalities. We aimed to establish additional molecular differences between HIF2A and non-HIF2A pseudo-hypoxic PGLs. We compared RNA expression patterns of six HIF2A PGLs from two patients with eight normal adrenal medullas and other hereditary pseudo-hypoxic PGLs (13 VHL, 15 SDHB, and 14 SDHD). Unsupervised hierarchical clustering showed that HIF2A PGLs made up a cluster that was separate from other pseudo-hypoxic PGLs. Significance analysis of microarray yielded 875 differentially expressed genes between HIF2A and other pseudo-hypoxic PGLs after normalization to the adrenal medulla. Prediction analysis of microarray allowed correct classification of all HIF2A samples based on as little as three genes—TRHDE (encoding thyrotropin-releasing hormone–degrading enzyme), LRRC63 (encoding leucine-rich repeat–containing protein 63), and IGSF10 (encoding immunoglobulin superfamily member 10). We selected genes with the highest expression difference between normal medulla and HIF2A PGLs for confirmatory quantitative reverse transcriptase polymerase chain reaction. In conclusion, HIF2A PGLs show a characteristic expression signature that separates them from non-HIF2A pseudo-hypoxic PGLs. Unexpectedly, the most significantly differentially expressed genes had not been previously described as HIF target genes.
Gangliocytic PGL (GPGL), a mixed neuro-ectodermal–endodermal tumor, is a rare and unique type of PGL that is almost exclusively located in the second portion of the duodenum, much like HIF2A–related somatostatinomas. Both GPGLs and HIF2A–related somatostatinomas produce and/or secrete somatostatin. In our series of 10 GPGLs, we found that two had HIF2A gene mutations.
Imaging of pheochromocytomas and paragangliomas
Patients with succinate dehydrogenase subunit B (SDHB) mutation–related PPGL are at a higher risk for metastatic disease than those with other hereditary PPGLs. Current therapeutic approaches are limited, but the best outcomes are based on the early and proper detection of as many lesions as possible. Because PPGLs overexpress somatostatin receptor 2 (SSTR2), the goal of our study was to assess the clinical utility of 68Ga-DOTATATE positron emission tomography/computed tomography (PET/CT) and to evaluate its diagnostic utility in comparison with the currently recommended functional imaging modalities 18F-fluorodopamine (18F-FDA), 18F-FDOPA, 18F-fluoro-2-deoxy-d-glucose (18F-FDG) PET/CT as well as CT/MRI. 68Ga-DOTATATE PET/CT was prospectively performed in 17 patients with SDHB–related metastatic PPGLs. All patients also underwent 18F-FDG PET/CT and CT/MRI, with 16 of the 17 patients also receiving 18F-FDOPA and 18F-FDA PET/CT scans. We compared detection rates of metastatic lesions among all these functional imaging studies. A composite synthesis of all used functional and anatomical imaging studies served as the imaging comparator. 68Ga-DOTATATE PET/CT demonstrated a lesion-based detection rate of 96.5–99.5%, 18F-FDG, 18F-FDOPA, 18F-FDA PET/CT, and CT/MRI showed detection rates of 81.3–89.4%, 55.6–66.9%, 46.1–57.7%, and 80.0–88.5%, respectively. We concluded, that 68Ga-DOTATATE PET/CT gives a detection rate significantly superior to all other functional and anatomical imaging modalities and may represent the preferred future imaging modality for evaluating SDHB–related metastatic PPGL.
Another prospective study included 22 patients (15 men, 7 women; aged 50.0 ± 13.9 years) with confirmed metastatic PPGL, a negative family history for PPGL, and negative genetic testing, who underwent 68Ga-DOTATATE, 18F-FDG PET/CT, and CT/MRI. Twelve patients underwent an additional 18F-FDOPA PET/CT scan and eleven an additional 18F-FDA PET/CT scan. We compared the rates of detection of metastatic lesions among all the imaging studies. A composite of all functional and anatomical imaging studies served as the imaging comparator. 68Ga-DOTATATE PET/CT showed a lesion-based detection rate of 95.8–98.7%. 18F-FDG PET/CT, 18F-FDOPA PET/CT, 18F-FDA PET/CT, and CT/MRI showed detection rates of 49.2%, 74.8%, 77.7%, and 81.6%, respectively. The results thus demonstrated the superiority of 68Ga-DOTATATE PET/CT in the localization of sporadic metastatic PPGLs compared with all other functional and anatomical imaging modalities (Figure 1) and suggest modification of future guidelines towards this new imaging modality. In the last study that included 20 patients with head and neck PGLs, we found that 68Ga-DOTATATE PET/CT was also superior to other imaging modalities.
Therapeutic aspects of pheochromocytoma and paraganglioma
Hypoxia is a common feature of solid tumors that activates a plethora of pathways, resulting in proliferation and resistance of cancer cells to radio- and chemotherapy. PPGLs with mutations in SDHB are the most aggressive forms of the disease, partly owing to their pseudo-hypoxic character, metabolic abnormalities, and elevated levels of reactive oxygen species (ROS). We investigated the effect of piperlongumine (PL), a natural product with cytotoxic properties towards cancer cells by significantly increasing intracellular ROS levels, on PHEO cells. We reported for the first time that PL mediates PHEO cell death by activating both apoptosis and necroptosis in vitro and in vivo. The effect is magnified in hypoxic conditions, making PL a promising potential candidate for use as a therapeutic option for patients with PPGL, including those with SDHB mutations.
In another study, we evaluated a novel topoisomerase I inhibitor, LMP-400, as a potential treatment for this devastating disease. We found high expression of topoisomerase I in human metastatic PHEO, thus providing a basis for the evaluation of a topoisomerase 1 inhibitor as a therapeutic strategy. LMP-400 inhibited the cell growth of established mouse PHEO cell lines and primary human tumor tissue cultures. In a study performed in athymic female mice, LMP-400 demonstrated a significant inhibitory effect on tumor growth with two drug administration regimens. Furthermore, low doses of LMP-400 lowered the protein levels of hypoxia-inducible factor 1 (HIF-1α), one of a family of factors studied as potential metastatic drivers in these tumors. The HIF-1α decrease resulted in changes in the mRNA levels of HIF-1 transcriptional targets. In vitro, LMP-400 showed an increase in the growth-inhibitory effects in combination with other chemotherapeutic drugs that are currently used for the treatment of PHEO. We concluded that LMP-400 had promising antitumor activity in preclinical models of metastatic PHEO and that its use should be considered in future clinical trial.
F1FoATP synthase (ATP synthase) is a ubiquitous enzyme complex in eukaryotes. In general, it is localized in the mitochondrial inner membrane and serves as the last step in the mitochondrial oxidative phosphorylation of ADP to ATP, utilizing a proton gradient across the inner mitochondrial membrane built by the complexes of the electron transfer chain. However, some cell types, including tumors, carry ATP synthase on the cell surface. It has been suggested that cell-surface ATP synthase helps tumor cells thriving on glycolysis survive their high acid generation. Angiostatin, aurovertin, resveratrol, and antibodies against the α and β subunits of ATP synthase were shown to bind to and selectively inhibit cell surface ATP synthase, promoting tumor cell death. We showed that ATP synthase β (ATP5B) is present on the cell surface of mouse PHEO cells as well as tumor cells of human SDHB–derived PGLs, while being virtually absent from chromaffin primary cells of bovine adrenal medulla, as shown by confocal microscopy. Using immuno–electron microscopy, we verified the cell-surface location of ATP5B in the tissue of an SDHB–derived PGL. Treatment of mouse PHEO cells with resveratrol as well as ATP5B antibody led to statistically significant inhibition of proliferation. Our data suggest that PGLs carry ATP synthase on their surface that promotes cell survival or proliferation. Thus, cell-surface ATP synthase may present a novel therapeutic target in treating metastatic or inoperable PGLs.
Animal model of pheochromocytoma and cell culture studies
A major impediment to the development of effective treatments for metastatic or unresectable PPGL has been the absence of valid models for pre-clinical testing. Attempts to establish cell lines or xenografts from human PPGLs have previously been unsuccessful. NOD-scid gamma (NSG) mice are a recently developed strain lacking functional B cells, T cells, and NK cells. We found that, in NSG mice, xenografts of primary human PGLs took, while maintaining the architectural and immuno-phenotypic characteristics expressed in the patients. In contrast to grafts of cell lines and of most common types of primary tumors, the growth rate of grafted PGLs is very slow, accurately representing the growth rate of most PPGLs, even in human metastases. Although the model is therefore technically challenging, primary patient-derived xenografts of PGLs in NSG mice provide a potentially important new tool that could prove especially valuable for testing treatments aimed at eradicating the small tumor deposits that are often numerous in patients with metastatic PGL.
Publications
- Darr R, Nambuba J, Del Rivero J, Janssen I, Merino M, Todorovic M, Balint B, Jochmanova I, Prchal JT, Lechan R, Tischler AS, Popovic V, Miljic D, Adams KT, Prall FR, Ling A, Golomb MR, Ferguson M, Nilubol N, Chen C, Chew E, Taieb D, Stratakis CA, Fojo A, Yang C, Kebebew E, Zhuang Z, Pacak K. Novel insights into the polycythemia-paraganglioma-somatostatinoma syndrome. Endocr Relat Cancer 2016;23(12):899-908.
- Turkova H, Prodanov T, Maly M, Martucci V, Adams K, Widimsky J Jr, Chen CC, Ling A, Kebebew E, Stratakis CA, Fojo T, Pacak K. Characteristics and outcomes of metastatic and sporadic pheochromocytoma/paraganglioma: a National Institutes of Health study. Endocr Practice 2016;22:302-314.
- Schovanek J, Bullova P, Tayem Y, Giubellino A, Wesley R, Lendvai N, Nölting S, Kopacek J, Frysak Z, Pommier Y, Kummar S, Pacak K. Inhibitory effect of the noncamptothecin topoisomerase I inhibitor LMP-400 on female mice models and human pheochromocytoma cells. Endocrinology 2015;156:4094-4104.
- Janssen I, Blanchet EM, Adams K, Chen CC, Millo CM, Herscovitch P, Taieb D, Kebebew E, Lehnert H, Fojo AT, Pacak K. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2016;21:3888-3895.
- Fliedner SM, Shankavaram U, Marzouca G, Elkahloun A, Jochmanova I, Daerr R, Linehan WM, Timmers H, Tischler AS, Papaspyrou K, Brieger J, de Krijger R, Breza J, Eisenhofer G, Zhuang Z, Lehnert H, Pacak K. Hypoxia-inducible factor 2a mutation-related paragangliomas classify as discrete pseudohypoxic subcluster. Neoplasia 2016;18(9):567-576.
Collaborators
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Alessio Imperiale, MD, PhD, Hôpital de Hautepierre, Les Hôpitaux Universitaires de Strasbourg, Strasbourg, France
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston, MA
- Jacques Lenders, MD, Radboud Universiteit, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Margarita Raygada, PhD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- David Taieb, MD, PhD, Centre Européen de Recherche en Imagerie Médicale, Hôpital de la Timone, Marseille, France
- Henri Timmers, MD, PhD, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, Clinical Center, NIH, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit http://pheopara.nichd.nih.gov.