Molecular Genetics of Endocrine Tumors and Related Disorders

- Constantine Stratakis, MD, D(med)Sci, Head, Section on Endocrinology and Genetics
- Maya Lodish, MD, Staff Clinician
- Fabio Faucz, PhD, Visiting Scientist
- Eva Szarek, BS, Visiting Scientist
- Edra London, PhD, Research Fellow
- Charalampos Lyssikatos, MD, Research Associate
- Annabel Berthon, PhD, Visiting Fellow
- Angela Dimopoulou, MD, Visiting Fellow (DIPHR)
- Paraskevi Salpea, PhD, Visiting Fellow
- Giampaolo Trivellin, PhD, Visiting Fellow
- Margarita Raygada, PhD, Genetic Counselor NICHD-NHGRI
- Elena Belyavskaya, MD, PhD, Physician Assistant
- Mihail Zilbermint, MD, Clinician Associate (Johns Hopkins University)
- Madelauz (Lucy) Sierra, MSc, Research Assistant
- Jenna Shapiro, BS, Graduate Student
- Smita Abraham (Baid), MD, Volunteer (FDA)
- Leticia Ferro Leal, BSc, Graduate Student
- Aaron Hodes, BSc, Adjunct Researcher
- Paola Chrysostomou, Adjunct Researcher
- Andreas Giannakou, MD, Adjunct Researcher
- Ludivine Drougat, PhD, Visiting Fellow
- Andrea Maria Gutierez, BS, Visiting Student (Brazil)
- Fady Hannah-Shmouni, MD, Clinical Associate (Endocrinology Fellow)
- Laura Hernandez-Ramirez, MD, PhD, Visiting Fellow
- Nickolas Settas, PhD, Visiting Fellow
- Amit Tirosh, MD, Contractor - Clinical Scientist (Israel)
- Christina Tatsi, MD, Clinical Associate (Pediatric Endocrinology Fellow)
- Jennifer Gourgaris, MD, Guest Researcher (Georgetown University faculty)
- Flora Bacopoulou, MD, Clinical Researcher on Sabbatical (Greece)
- Pablo Beckers, MD, Clinical Researcher on Rotation (Belgium)
- Stephanie Espiard, MD, Visiting Fellow on Grant (France)
- Nuria Valdes Gallego, MD, Clinical Researcher on Sabbatical (Spain)
- Lloyd Tack, MD, Visiting Clinician on Grant (Belgium)
- Marianna Athanassiou, Summer Student
- Hailey Blain, Summer Student
- Rebecca Boden, Summer Student
- Christine Dedoulis, Summer Student
- Maria Faidas, Summer Student
- Mia Faulkner, Summer Student
- Eleanor Kimmelman, Summer Student
- Gregory Papaioannou, Summer Student
- Paulina Pedas, Summer Student

Click image to enlarge.
SEGEN Laboratory Staff and Dr. Stratakis
Our staff and Dr. Constantine Stratakis in March 2016 at our laboratory at the NIH CRC (1-East-3216)
The project “Molecular Genetics of Endocrine Tumors and Related Disorders” was started in the late 1990s. The goal has always been to identify molecular pathways involved in the first steps of tumor formation. Our approach was to study patients with rare endocrine conditions, mostly inherited, identify the causative genes, and then study the involved signaling pathways in the hope of translating the derived knowledge into new therapies for such patients. The derived knowledge could also be generalized to conditions that are not necessarily inherited, e.g., to more common tumors and diseases caused by defects in the same molecular pathways. The approach has indeed led to fruitful research over the last two decades.
Our first studies led to the identification of the main regulator of the cAMP signaling pathway, the regulatory subunit–type 1A (R1a) of protein kinase A (PKA, encoded by the PRKAR1A gene on chromosome 17q22-24), as responsible for primary pigmented nodular adrenocortical disease (PPNAD) and the Carney complex, a multiple endocrine neoplasia (MEN), whose main endocrine manifestation is PPNAD. We then focused on clinically delineating the various types of primary bilateral adrenal hyperplasias (BAH). We described isolated micronodular adrenocortical disease (iMAD), a disorder likely to be inherited in an autosomal dominant manner and unrelated to the Carney complex or to other MENs. The identification of PRKAR1A mutations in PPNAD led to the recognition that non-pigmented forms of BAHs existed, and a new nomenclature was proposed, which we first suggested in 2008 and has since become used worldwide.
In 2006, a genome-wide association (GWA) study led to the identification of mutations in the phosphodiesterases (PDE) PDE11A, a dual specificity PDE, and PDE8B, a cAMP–specific PDE (encoded by the PDE11A and PDE8B genes, respectively) in iMAD. Following the establishment of cAMP/PKA involvement in PPNAD and iMAD, we and others found that elevated cAMP levels and/or PKA activity and abnormal PDE activity may be found in most benign adrenal tumors (ADTs), including the common adrenocortical adenoma (ADA). We then found PDE11A and PDE8B mutations or functional variants in adrenocortical cancer (ACA) and in other forms of adrenal hyperplasia such as massive macronodular adrenocortical disease (MMAD), also known as ACTH–independent adrenocortical hyperplasia (MMAD/AIMAH). Germline PDE11A sequence variants may also predispose to testicular cancer (testicular germ cell tumors or TGCTs) and prostate cancer, indicating a wider role of this pathway in tumor formation on cAMP–responsive, steroidogenic, or related tissues. Ongoing work with collaborating NCI laboratories aims to clarify the role of PDE in predisposition to these tumors. It is clear from these data, however, that there is significant pleiotropy of PDE11A and PDE8B defects. The histo-morphological studies that we performed on human adrenocortical tissues from patients with these mutations showed that iMAD is highly heterogeneous and thus likely to be caused by defects in various genes of the cAMP/PKA signaling pathway or its regulators and/or downstream effectors.
Similarly, the G protein–coupled receptor (GPCR)–linked MMAD/AIMAH disease includes a range of adrenal phenotypes from very similar to iMAD to the GNAS–caused primary bimorphic adrenocortical disease (PBAD) and McCune-Albright syndrome, caused by somatic mutations of the GNAS gene (encoding the G protein–stimulatory subunit alpha or Gsa). Although a few of the patients with MMAD/AIMAH have germline PDE11A, PDE8B, or somatic GNAS mutations, others have germline fumarate hydratase (FH), menin (MEN1), and adenomatous polyposis coli (APC) mutations, pointing to the range of possibilities and the pathways that may be involved. Particularly interesting among these are FH mutations associated with mitochondrial oxidation defects that have been linked to adrenomedullary tumors. This led us to investigate a disorder known as the Carney Triad, the only known disease that has among its clinical manifestations both adrenocortical (ADA, MMAD/AIMAH) and medullary tumors (pheochromocytomas [PHEOs] and paragangliomas [PGLs]), in addition to hamartomatous lesions in various organs (pulmonary chondromas and pigmented and other skin lesions) and a predisposition to gastrointestinal stromal tumors or sarcomas (GISTs). A subgroup of patients with PHEOs, PGLs, and GISTs were identified as harboring mutations in succinate dehydrogenase (SDH) subunits B, C, and D (encoded by the SDHB, SDHC, and SDHD genes, respectively); the patients also rarely have adrenocortical lesions, ADAs, and/or hyperplasia, and their disease is known as the dyad or syndrome of PGLs and GISTs or, as named by a group of pathologists and now in wide use, the Carney-Stratakis syndrome (CSS).
In 2013, MMAD/AIMAH was renamed primary macronodular adrenocortical hyperplasia (PMAH) after it was discovered that it depends on adrenoglandular ACTH production, at least occasionally. As part of this work, a new gene was identified (ARMC5) that, when mutated, causes more than a third of the known PMAH cases. The function of the gene is unknown, and we have embarked on a project to characterize it further, including studying mouse, fruit fly, and fish models. The ARMC5 gene has a beta-catenin–like motif.
PPNAD appears to be less heterogeneous and is mostly caused by PRKAR1A mutations, but up to one third of patients with the classic features of PPNAD do not have PRKAR1A mutations, deletions, or 17q22-24 copy-number variant (CNV) abnormalities. A subset of these patients may have defects in other molecules of the PKA holoenzyme, and their study is important for understanding how PKA works and the tissue specificity of each defect. For patients with disorders that are yet to be elucidated on a molecular level, we continue to delineate the phenotypes and identify the responsible genetic defects through a combination of genomic and transcriptomic analyses.
Recently, we identified genes encoding two other subunits of PKA as involved in endocrine tumors: PRKACA in BAH and PRKACB in a form of Carney complex that is not associated with PRKAR1A mutations. Our laboratory is now investigating these two genes.
Animal model studies are essential for the investigation and confirmation of each of the identified new genes in disease pathogenesis. Furthermore, such studies provide insight into function that can be tested quickly in human samples for confirmation of its relevance to human disease. One excellent example of such a bench-to-bedside (and back) process in the last review cycle was the identification, from a variety of animal experiments, of Wingless/int (Wnt) signaling as one of the downstream effectors of tumor formation in the context of increased cAMP/PKA activity. Both our laboratory and our collaborators found somatic beta-catenin (CTNNB1) mutations in large ADAs that formed in the background of PPNAD caused by germline PRKAR1A mutations. Our transcriptomic studies had previously identified the WNT1–inducible signaling pathway protein 2 (WISP2) as the main molecule overexpressed in food-dependent Cushing’s syndrome caused by MMAD/AIMAH, and our recent micro-RNA studies showed that genes that regulate WNT signaling were major targets of micro-RNAs, which were found dysregulated in both PPNAD and MMAD/AIMAH. Cells from tumors or other lesions from animals with R1a deficiency showed elevated beta-catenin expression and/or aberrant WNT signaling and similarities to adult stem cells or cancer stem cells in other models of dysregulated WNT signaling. However, it appears that beta-catenin activation in R1a–deficient cells is preceded by yet unknown molecular abnormalities that take place within the still benign and R1a–haploinsufficient tissues in the early stages of tumor formation.
We continue to investigate the pathways involved in early events in tumor formation in the adrenal cortex and/or the tissues affected by germline or somatic defects of the cAMP/PKA and related endocrine signaling defects, employing animal models and transcriptomic and systems-biology analyses. Understanding the role of the other PKA subunits in this process is essential. An example of the combined use of whole genomic tools, transcriptomic analysis, and mouse and zebrafish models to investigate the function of a gene or a pathway is the ongoing work on the Carney Triad.
An important discovery in the last 3–4 years was that mice with neural crest–, heart-, and adrenal-specific knockouts (KO) of R1a or mice with other R1a defects develop lesions caused by proliferation of stem cell–like, tissue-specific pluripotent cells (TSPCs) in adult tissues such as the adult skeleton. We studied bone and the adrenal cortex. Given that various models of R1a deficiency appear to feature the growth of lesions derived from TSPCs, we are characterizing these cells in the adrenal and bone and creating laboratory conditions (i.e., culture systems) to propagate them in vitro, study their growth and proliferation, exploit their therapeutic potential, and/or identify molecules that affect the cells for targeting the related tumors in humans.
We continue to accrue patients under several clinical protocols, identify unique patients and families with rare phenotypes, and/or explore (mostly on a collaborative basis) various aspects of endocrine and related diseases. Paramount to these investigations is the availability of modern genetic tools such as copy number variation (CNV) analysis, comparative genomic hybridization (CGH), whole-exome sequencing (WES), and DNA sequencing (DSeq). As part of these clinical protocols, much clinical research is also being done, consisting mostly of observations of new associations, description of novel applications or modifications, and improvements in older diagnostic methods, tests, or imaging tools. This is a particularly fruitful area of research, especially for our clinical fellows who matriculate at our laboratory during their two-year research time. The approach also leads to important new discoveries that may steer us into new directions.
One such discovery was the recent identification by our laboratory of the defect that explains the vast majority of cases of early pediatric overgrowth or gigantism. What regulates growth, puberty, and appetite in children and adults is poorly understood. We identified the gene GPR101, encoding a G protein–coupled receptor, which was overexpressed in patients with elevated growth hormone (GH). Patients with GPR101 defects have a condition that we called X-LAG for X-linked acrogigantism caused by Xq26.3 genomic duplication and characterized by early-onset gigantism resulting from excess GPR101 function and consequently elevated GH.
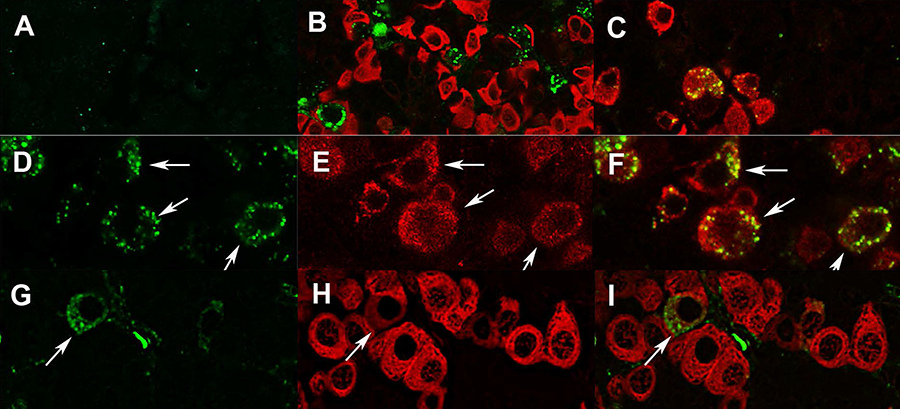
Click image to enlarge.
GPR101 expression in the pituitary and hypothalamus (Trivellin G et al. J Mol Endocrinol 2016;57:97–111)
Labeling of GPR101 in the anterior pituitary gland of female rhesus monkey (A–F) and rat (G–I). (A) Preincubation of GPR101 antibody with blocking peptide, resulted in no staining. B) Punctuate GPR101 (green) labeling could not be attributed to somatotrophs (red). (C) GPR101 (green) is expressed by LHβ–positive cells (red). (D–F) High magnification images showing membranous and cytoplasmic distribution of GPR101 in monkey gonadotrophs, excluding nucleus. (G–I) GPR101 (green) is expressed by subpopulation of rat somatotrophs (red), as indicated by arrows. Scale bars: A–D: 20 µm; E: 10 µm.
Carney complex (CNC) genetics
We have collected families with CNC and related syndromes from several collaborating institutions worldwide. Through genetic linkage analysis, we identified loci harboring genes for CNC on chromosomes 2 (2p16) and 17 (17q22–24) and are currently searching for other possible loci for this genetically heterogeneous condition. With the application of state-of-the-art molecular cytogenetic techniques, we are investigating the participation of these currently identified genomic loci in the expression of the disease and have constructed a comprehensive genetic and physical map of the 2p16 chromosomal region for cloning the CNC–associated sequences from this region. Studies in cultured primary tumor cell lines (established from our patients) identified a region of genomic amplification in CNC tumors in the center of the map. The PRKAR1A gene on 17q22–24, the gene responsible for CNC in most cases of the disease, appears to undergo loss of heterozygosity in at least some CNC tumors. PRKAR1A is also the main regulatory subunit (subunit type 1-a) of PKA, a central signaling pathway for many cellular functions and hormonal responses. We have increased the number of CNC patients in genotype-phenotype correlation studies, which are expected to provide insight into the complex biochemical and molecular pathways regulated by PRKAR1A and PKA. We expect to identify new genes by ongoing genome-wide searches for patients and families who do not carry PRKAR1A mutations.
PRKAR1A, protein kinase A activity, and endocrine and other tumor development
We are investigating the functional and genetic consequences of PRKAR1A mutations in cell lines established from CNC patients and their tumors. We measure both cAMP and PKA activity in the cell lines, along with the expression of the other subunits of the PKA tetramer. In addition, we are seeking mutations of the PRKAR1A gene in sporadic endocrine and non-endocrine tumors (thyroid adenomas and carcinomas, adrenocortical adenomas and carcinomas, ovarian carcinomas, melanomas and other benign and malignant pigmented lesions, and myxomas in the heart and other sites)—mutations that would further establish the gene’s role as a general tumor suppressor. Many investigators within the NIH and around the world provide specimens on a collaborative basis.
Prkar1a+/– and related animal models
Several years ago we developed a Prkar1a knockout mouse floxed by a lox-P system for the purpose of generating, first, a novel Prkar1a+/– and, second, knockouts of the Prkar1a gene in a tissue-specific manner after crossing the new mouse model with mice expressing the cre protein in the adrenal cortex, anterior lobe of the pituitary, and thyroid gland (Kirschner et al. Cancer Res 2005;65:4506). The heterozygote mouse develops several tumors reminiscent of the equivalent human disease. We have now developed new crosses that show protein kinase A subunit involvement in additional phenotypes.
PRKAR1A, the cell cycle, and other signaling pathways
We work to identify PRKAR1A–interacting mitogenic and other growth-signaling pathways in cell lines expressing PRKAR1A constructs and/or mutations. Several genes that regulate PKA function and increase cAMP–dependent proliferation and related signals may be altered in the process of endocrine tumorigenesis initiated by a mutant PRKAR1A, a gene with important functions in the cell cycle and in chromosomal stability. Recently, we found an interaction with the mTOR pathway in both human and mouse cells with altered PKA function.
Phosphodiesterase (PDE) genes in endocrine and other tumors
In patients who did not exhibit CNC or have PRKAR1A mutations but presented with bilateral adrenal tumors similar to those in CNC, we found inactivating mutations of the PDE11A gene, which encodes phosphodiesterase-11A, an enzyme that regulates PKA in the normal physiologic state. Phosphodiesterase 11A is a member of a 22 gene–encoded family of proteins that break down cyclic nucleotides controlling PKA. PDE11A appears to act as a tumor suppressor such that tumors develop when its action is abolished. In what proved to be the first cases in which mutated PDE was observed in a genetic disorder predisposing to tumors, we found pediatric and adult patients with bilateral adrenal tumors. Recent data indicate that PDE11A sequence polymorphisms may be present in the general population. The finding that genetic alterations of such a major biochemical pathway may be associated with tumors in humans raises the reasonable hope that drugs that modify PKA and/or PDE activity may eventually be developed for use in both CNC patients and those with other, non-genetic, adrenal tumors—and perhaps other endocrine tumors.
After the identification of a patient with a PDE8B mutation and Cushing’s syndrome, additional evidence emerged for yet another cAMP–specific PDE to be involved in endocrine conditions.
We also studied both Pde11a and Pde8b animal models.
Genetic investigations into other adrenocortical diseases and related tumors
Through collaborations, we (1) apply general and pathway-specific microarrays to a variety of adrenocortical tumors, including single adenomas and MMAD, to identify genes with important functions in adrenal oncogenetics; (2) examine candidate genes for their roles in adrenocortical tumors and development; and (3) identify additional genes that play a role in inherited pituitary, adrenocortical, and related diseases.
Genetic investigations into pituitary tumors, X-LAG, other endocrine neoplasias, and related syndromes
In collaboration with several other investigators at the NIH and elsewhere, we are investigating the genetics of CNC– and adrenal-related endocrine tumors, including childhood pituitary tumors, related or unrelated to PRKAR1A mutations. As part of this work, we have identified novel genetic abnormalities.
We identified the gene GPR101 encoding an orphan G protein–coupled receptor (GPCR), which was overexpressed in patients with elevated growth hormone (GH) or gigantism. Patients with GPR101 defects have a condition that we called X-LAG, for X-linked acrogigantism, caused by Xq26.3 genomic duplication and characterized by early-onset gigantism resulting from excessive GPR101 function and consequent GH excess. To find additional patients with this disorder, we collaborated with a group at the University of Liège headed by Albert Beckers, but all the molecular work for the gene identification was carried out here at the NIH. We have found that the gene is expressed in areas of the brain that regulate growth and we identified a few small-molecule compounds that bind to GPR101 (unpublished). The data are now being used to identify more specific compounds that bind to GPR101.
Genetic investigations into the Carney Triad, other endocrine neoplasias, and related syndromes and into hereditary paragangliomas and related conditions
As part of a collaboration with other investigators at the NIH and elsewhere (including an international consortium organized by our laboratory), we are studying the genetics of a rare syndrome that predisposes to adrenal and other tumors, the Carney Triad, and related conditions (associated with gastrointestinal stromal tumors, or GIST). In the course of our work, we identified a patient with a new syndrome, known as the paraganglioma and gastrointestinal stromal tumor syndrome (or the Carney-Stratakis syndrome), for which we found mutations in the genes encoding succinate dehydrogenase (SDH) subunits A, B, C, and D. In another patient, we found a novel germline mutation in the PDFGRA gene. Most recently, in collaboration with a group in Germany, we identified an epigenetic defect (methylation of the SDHC gene) that may be used diagnostically to identify patients with the Carney Triad.
Clinical investigations into the diagnosis and treatment of adrenal and pituitary tumors
Patients with adrenal tumors and other types of Cushing’s syndrome (and occasionally other pituitary tumors) come to the NIH Clinical Center for diagnosis and treatment. Ongoing investigations focus on (1) the prevalence of ectopic hormone receptor expression in adrenal adenomas and PMAH/MMAD; (2) the diagnostic use of high-sensitivity magnetic resonance imaging for earlier detection of pituitary tumors; and (3) the diagnosis, management, and post-operative care of children with Cushing’s syndrome and other pituitary tumors.
Clinical and molecular investigations into other pediatric genetic syndromes
Mostly in collaboration with several other investigators at the NIH and elsewhere, we are conducting work on pediatric genetic syndromes seen in our clinics and wards.
Additional Funding
- INSERM, Paris, France (Co-Principal Investigator): “Clinical and molecular genetics of Carney complex,” 06/2003-present
- Several small grants supporting staff members from France, Brazil, Greece, Spain, and elsewhere
Publications
- Drelon C, Berthon A, Sahut-Barnola I, Mathieu M, Dumontet T, Rodriguez S, Batisse-Lignier M, Tabbal H, Tauveron I, Lefrançois-Martinez AM, Pointud JC, Gomez-Sanchez CE, Vainio S, Shan J, Sacco S, Schedl A, Stratakis CA, Martinez A, Val P. PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat Commun 2016;7:12751.
- Bandettini WP, Karageorgiadis AS, Sinaii N, Rosing DR, Sachdev V, Schernthaner-Reiter MH, Gourgari E, Papadakis GZ, Keil MF, Lyssikatos C, Carney JA, Arai AE, Lodish M, Stratakis CA. Growth hormone and risk for cardiac tumors in Carney complex. Endocr Relat Cancer 2016;23:739-746.
- Saloustros E, Liu S, Mertz EL, Bhattacharyya N, Starost MF, Salpea P, Nesterova M, Collins M, Leikin S, Stratakis CA. Celecoxib treatment of fibrous dysplasia (FD) in a human FD cell line and FD-like lesions in mice with protein kinase A (PKA) defects. Mol Cell Endocrinol 2016;439:165-174.
- Gourgari E, Lodish M, Keil M, Sinaii N, Turkbey E, Lyssikatos C, Nesterova M, de la Luz Sierra M, Xekouki P, Khurana D, Ten S, Dobs A, Stratakis CA. Bilateral adrenal hyperplasia as a possible mechanism for hyperandrogenism in women with polycystic ovary syndrome. J Clin Endocrinol Metab 2016;101:3353-3360.
- Trivellin G, Bjelobaba I, Daly AF, Larco DO, Palmeira L, Faucz FR, Thiry A, Leal LF, Rostomyan L, Quezado M, Schernthaner-Reiter MH, Janjic MM, Villa C, Wu TJ, Stojilkovic SS, Beckers A, Feldman B, Stratakis CA. Characterization of GPR101 transcript structure and expression patterns. J Mol Endocrinol 2016;57:97-111.
Collaborators
- Albert Beckers, MD, Université de Liège, Liège, Belgium
- Jerome Bertherat, MD, PhD, Service des Maladies Endocriniennes et Métaboliques, Hôpital Cochin, Paris, France
- Stephan Bornstein, MD, PhD, Universität Dresden, Dresden, Germany
- Isabelle Bourdeau, MD, Université de Montréal, Montreal, Canada
- J. Aidan Carney, MD, PhD, Mayo Clinic, Rochester, MN
- Nickolas Courkoutsakis, MD, PhD, University of Thrace, Alexandroupolis, Greece
- Jennifer Gourgari, MD, Georgetown University, Washington, DC
- Margaret F. Keil, PhD, RN, CRNP, Office of the Clinical Director, NICHD, Bethesda, MD
- Lawrence Kirschner, MD, PhD, James Cancer Hospital, Ohio State University, Columbus, OH
- Andre Lacroix, MD, PhD, Centre Hospitalier de l’Université de Montréal, Montreal, Canada
- Giorgios Papadakis, MD, MPH, Nuclear Medicine, Clinical Center, NIH, Bethesda, MD
- Nickolas Patronas, MD, Diagnostic Radiology, Clinical Center, NIH, Bethesda, MD
- Margarita Rayada, PhD, Office of the Clinical Director, NICHD, Bethesda, MD
Contact
For more information, email stratakc@mail.nih.gov or visit http://segen.nichd.nih.gov.